Services Test Online
PDISORDER: The Program for Finding Intrinsic Disorder Regions in Protein Sequences
PDISORDER V. 1.0 is the program for predicting ordered and disordered regions in protein sequences. Minimum required sequence length is 40.
It is increasingly evident that intrinsically unstructured protein regions play key roles in cell-signaling, regulation and cancer (Iakoucheva et al., J. Mol. Biol. (2002) 323, 573�584), which makes them extremely useful for discovery of anticancer drugs. Requirement of intrinsic structural disorder is shown for many protein functions - see, for instance, Dunker et al., Biochemistry (2002) 41 (21), 6573 -6582.
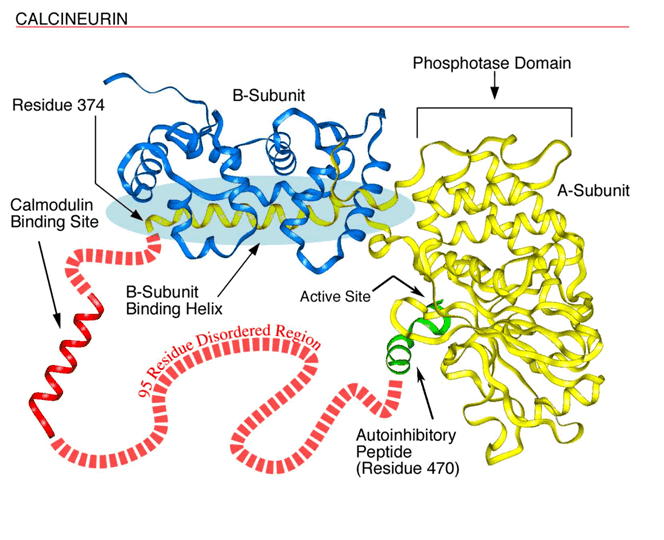
The figure below shows disorderly region in Calcineurin (reproduced from ORNL Human Genome News), see output example below for prediction of its disorder region.
Combination of Neural Network, Linear Discriminant Function and acute Smoothing Procedure is used for recognition of disordered and ordered regions in proteins.
Two sets of significant attributes: one for Neural Network, and another one for Linear Discriminant Function are selected using automatic LDA procedure, as well as approach based on calculations of chances to be in disordered or ordered regions.
Three windowing procedures are used, called left, right and intermediate. For all windows, attributes are calculated over 31 residues.
Example of PDISORDER output:
Prediction of disordered regions in proteins. Softberry Inc.
>gi|1352677|sp|P48457|P2B_EMENI Ser/thr protein phosphatase 2B catalytic subunit
Calmodulin-dependent calcineurin A subunit)
10 20 30 40
Pred_od ooooooooo ddd ooooooooooooooooooooooooooooooooo
AA seq MEDGTQVSTLERVVKEVQAPALNKPSDDQFWDPEEPTKPNLQFLKQHFYR
Prob_o 66666665655663335777766565589767999999999999997999
60 70 80 90
Pred_od oooooooooooooooooooooooooooooooooooooooooooooooooo
AA seq EGRLTEDQALWIIQAGTQILKSEPNLLEMDAPITVCGDVHGQYYDLMKLF
Prob_o 99999999999999999999999999999999999999999999999999
110 120 130 140
Pred_od oooooooooooooooooooooooooooooooooooooooooooooooooo
AA seq EVGGDPAETRYLFLGDYVDRGYFSIECVLYLWALKIWYPNTLWLLRGNHE
Prob_o 99999999999999999999999999999999999999999999999999
160 170 180 190
Pred_od oooooooooooooooooooooooooooooooooooooooooooooooooo
AA seq CRHLTDYFTFKLECKHKYSERIYEACIESFCALPLAAVMNKQFLCIHGGL
Prob_o 99999999999999999999999999999999999997555556887888
210 220 230 240
Pred_od oooooooooooooooooooooooooooooooooooooooooooooooooo
AA seq SPELHTLEDIKSIDRFREPPTHGLMCDILWADPLEDFGQEKTGDYFIHNS
Prob_o 78775555553563478776666666678689999999999999999999
260 270 280 290
Pred_od oooooooooooooooooooooooooooooooooooooooooooooooooo
AA seq VRGCSYFFSYPAACAFLEKNNLLSVIRAHEAQDAGYRMYRKTRTTGFPSV
Prob_o 99999999999999999999999999999999999999999999999999
310 320 330 340
Pred_od oooooooooooooooooooooooooooooooooooooooooooooooooo
AA seq MTIFSAPNYLDVYNNKAAVLKYENNVMNIRQFNCTPHPYWLPNFMDVFTW
Prob_o 99999999999999999999999999999999999999999999999999
360 370 380 390
Pred_od ooooooooooo dddddddddddddddddddddddddddd
AA seq SLPFVGEKITDIVIAILNTCSKEELEDETPSTISPAEPSPPMPMDTVDTE
Prob_o 99999976656555554444441100000000000000000000000000
410 420 430 440
Pred_od dddddddddddddddddddddddddddddddddddddddddddddddddd
AA seq STEFKRRAIKNKILAIGRLSRVFQVLREESERVTELKTAAGGRLPAGTLM
Prob_o 00000000000100000000001223333444444333422232555555
460 470 480 490
Pred_od dddddddddddddddddddddddddddddddddddddddddddddddddd
AA seq LGAEGIKQAITNFEDARKVDLQNERLPPSHDEVVRRSEEERRIALDRAQH
Prob_o 55555433255544555565443400000231112100000000000001
510 520
Pred_od dddddddddddddddddddddddddddddd
AA seq EADNDTGLATVARRISMVRRIRKIPSTTRR
Prob_o 020000022332232444444444443343
sequences=1 disordered=161 ordered=353 unknown=16
Here line Pred_od shows ordered (o) and disordered (d) regions. Blanks denote undefined-state stretches, usually at boundaries of disordered regions.
Line Prob_o shows raw probability on a scale of 0 to 9 for each amino acid residue to be in ordered region.
The line at the end of the output shows total number of sequence residues in each state: disordered, ordered and unknown.
Accuracy estimations:
One of accuracy tests was made on PONDR data and in comparison with PONDR.
Black and blue - PONDR's data, green - our descriptions, red - PDISORDER results.
PONDR and PDISORDER accuracies
| Predictor | False Negative (dis_ALL) - 124 sequences >31 in lengths, 17181 positions (false, true) | False Positive (O_PDB_S25) - 1081 sequences >31 in lengths, 220743 positions (false, true) | 5-cross Validation | Unknown (for both sets) |
||
| VL-XT | 40% | - | 22% | - | 75 - 83% | - |
| XL1 | 62% | - | 19% | - | 73 ± 4% | - |
| CaN | 39% | - | 34% | - | 83 ± 5% | - |
| PDISORDER | 20.3% | 78.3% | 4.7% | 94.4% | - | 0.7% |