It is believed that most miRNAs are scarce in the cell and therefore are not yet discovered. The program FindMiRNA searches for miRNA genes and miRNAs within them.
The search procedure
The search process is conducted by successive filtering the genomic sequence.
The procedure is organized in four steps: 1) fast estimation of secondary structure
potential by calculation nucleotide scores; 2) search for hairpins and calculation
of their energies; 3) estimation of thermodynamic probability of the hairpin
structure found; 4) search for miRNAs in the candidate hairpin. In more details these
filters are described below.
At first the FindMiRNA scans the input sequence with the sliding window of 100nt. Within the window it calculates nucleotide content and estimates E-score (the sequence potential to form stable secondary structure). It filters out the subsequences can not form the stable stable structures, i.e. which nucleotide content and E-score don not fall in the range of found miRNA genes. For clever filtering it takes into account the interdependency of nucleotide scores and interdependency of overlapping sequence windows. The step is the fastest one with time complexity of O(N).
At the second step FindMiRNA calls for another Softberry program, BestPal, which calculates the optimal imperfect hairpin which can be formed within a sequence window. The BestPal algorithm is based on the idea of dynamic programming realized in the wide-spread mfold algorithm for RNA secondary structure prediction. BestPal uses the energy parameters of Turner's energy rules. The hairpin energy is calculated summing over the energies of helixes and loops:
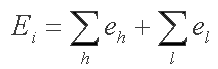
where eh is helix energy and el is loop energy.
Searching for hairpins, BestPal omits secondary structure junctions and therefore works faster than Zuker's mfold program. Its time complexity is O(N2.88) comparing with O(N3.5) of mfold. When BestPal work is completed, the FindMiRNA saves the subsequences with stable hairpins only (free energy less than -17 kcal/mole by default). Though it takes most time, currently this step is the most effective in reducing the pre-miRNA candidate number.
At the third step FindMiRNA calls for RNAfold_bpp program. This filter takes the remaining sequences and calculates their matrices of base-pairing probabilities. The algorithm is based on McCaskill algorithm and dynamically calculates the partition function of RNA. Using partition function, our program calculates base-pairing probabilities of the ensemble of RNA structures. Using the optimal hairpin structure calculated at step 2, it estimates the hairpin probability and filters out the sequences with stable alternative structures. This step has the slowest time complexity of O(N3.5), however, the initial sequence is already reduced by several orders at the steps 1 and 2.
At the final step FindMiRNA searches for miRNAs within the sequences remained. It calculates the weight matrix of any 21-mer oligonucleotide within a putative pre-miRNA and takes into account base-pairing characteristics of a candidate miRNA.
Currently the program is specially trained for three organisms (hsa, mmu and ath), although it can be used for others. We plan to extend the number of organisms analyzed and to automatically detect which of the analyzed genomes an input sequence belongs to.
Input and output
The program input is a genomic sequence and three-letter organism ID.
The program outputs the putative pre-miRNAs and miRNAs in the following order:
- chain direction (+\-)
- the beginning and the end of a predicted pre-miRNA
- the beginning and the end of a predicted miRNA
- pre-miRNA sequence
- miRNA sequence