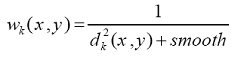 ,
,
Normalization of the Affymetrix gene expression row data by MAS 5.0 algorithm.
Data specification
The input for MAS5Norm is the set of expression row data in Affymetrix CEL data format, corresponding CDF file and file with list of CEL files to be processed ant their short description (this file is provided by user). The CEL file stores the results of the intensity calculations on the pixel values on the chip. The CDF file describes the layout for an Affymetrix GeneChip array. The output is SelTag data file with gene expression data.
Algorithm description
The purpose of the algorithm is to subtract background noise from the row probe intensities on the chip and perform data normalization to obtain normalized and scaled signal values for gene expression. The method is known as MAS 5.0 statistical algorithm implemented in the Affymetrix Microarray Suite version 5.0. The algorithm details are described in the Affymetrix documentation at http://www.affymetrix.com/support/technical/technotesmain.affx ("Statistical Algorithms Description Document", Affymetrix, 2002; "Statistical Algorithms Reference Guide", Affymetrix, 2001).
Background noise correction. At the first step the chip area is divided into K squared zones of the same size (default number of zones is 16). Then the 2% probes with the lowest intensity define the background intensity for each zone. The background noise level for each k-th zone bZk is the calculated as the average for those lowest intensity probes. The background noise level b(x,y) for each probe at the chip location x,y is calculated as weighted sum of zone background values
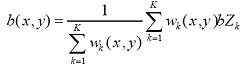 ,
,
where weights wk(x,y) are calculated as follows:
,
where dk(x,y) is the distance from the point x,y to the center of the k-th zone, smooth - is the smoothing parameter (by default is 100).
The noise correction procedure is as follows. First, standard deviations of the 2% probes with the lowest intensity nZk are calculated for each zone. For each probe the noise intensity n(x,y) is is estimated by above formulas (substitute n(x,y) for b(x,y) and nZk for bZk in the formulas above). Then the probe intensity corrected for noise is calculated from actual probe intensity I(x,y) as follows:
A(x,y)=max(I'(x,y)-b(x,y),NoiseFrac*n(x,y)),
where I'(x,y)=max(I(x,y),0.5), NoiseFrac is the fraction of noise and is set to 0.5 as in MAS 5.0 algorithm description.
Expression value (signal) calculation. After background subtraction from each probe intensity value, the signal values for the probesets are calculated. The calculation uses "ideal mismatch" technique that allows to process probe pairs for which the mismatch (MM) signal is greater than the match (PM) signal (see details in the Affymetrix documentation). When the ideal mismatch is calculated for each probe pair j of the each probeset i, the probe value PVij is calculated: PVij = log2(max(PMij-IMij, 2-20)). The signal log value (SLVi) for the probeset i is calculated as the one-step biweight estimate for the corresponding probeset SLVs. Then the algorithm scales all the probesets to target scale value Sc (default is 500) estimating the scale factor sf
and using normalization factor nf (for this program is always set to 1):
Signal=sf·nf·2SLVi. The TrimMean function calculates t he mean value of the data without highest 2% and lowest 2% values.
Estimation of the signal statistical significance (detection p-values). To estimate the significance of the signal deviation from noise Wilcoxon's rank test is used. This test determines the significance of the deviation of the discrimination score Ri for the probeset i
from the threshold value t (this value specified by user, by default is set to 0.015). The significance of the deviation of the Ri from t is calculated by Wilcoxon's rank test and reported as detection p-value.
Estimation of the presence/absence of the signal (detection call). The algorithm report three types of detection calls: present (P), marginal detection (M) or absent (A). The detection is based on the p-value and two user-defined parameters, a1 and a2: the signal is present if p< a1; the signal is marginally present if a1<p<a2. The signal is absent if p<2. By default a1 = 0.04 and a2 = 0.06 (for 16-20 probe pairs).
The program can analyze a set of CEL data files corresponding for the same CDF chip data. The output file is in SelTag format and reports the #HEADER section: Chip name; for each experiment (CEL file) ExperimentDataFilename, DataHeader as reported in the user-defined CEL list file, DataScalingFactor (sf value), DataNormalizationFactor (nf value), DataSignalTrimmedMean.
Example of experiment list file
GSM42890 DEHP_48hr_Veh1 DEHP 48hr Veh1 GSM42891 DEHP_48hr_Veh2 DEHP 48hr Veh2 GSM42892 DEHP_48hr_Veh3 DEHP 48hr Veh3 GSM42893 DEHP_48hr_Veh4 DEHP 48hr Veh4 GSM42894 DEHP_48hr_Veh5 DEHP 48hr Veh5
This file contains three columns separated by
Example of output data
#HEADER Multiple chip data analysis by Affymetrix MAS5.0 algoritm. ChipName=RG_U34A. 1 ExperimentDataFilename=GSM42890.cel 1 DataHeader=DEHP_48hr_Veh1 DEHP 48hr Veh1 1 DataScalingFactor=7.4530 1 DataNormalizationFactor=1.0000 1 DataSignalTrimmedMean=1500.0000 MAS5 algorithm parameters: BF=2.0000 NZ=16 Bsmooth=100.0000 Alpha1=0.0400 Alpha2=0.0600 TGT=1500.0000 #ENDHEADER ProbesetName STRING DEHP_48hr_Veh1_Signal FVALUE DEHP_48hr_Veh1_Detection WORD DEHP_48hr_Veh1_Detection_p FVALUE END DATA AFFX-MurIL2_at 37.5396 A 0.78955 AFFX-MurIL10_at 51.8929 A 0.60308 AFFX-MurIL4_at 5.7568 A 0.97607 AFFX-MurFAS_at 32.2922 A 0.60308 AFFX-BioB-5_at 714.0201 A 0.08359 AFFX-BioB-M_at 1563.2017 P 0.00125 AFFX-BioB-3_at 800.5414 P 0.00359 AFFX-BioC-5_at 3686.6155 P 0.00017 AFFX-BioC-3_at 1989.3492 P 0.00006 AFFX-BioDn-5_at 2807.6296 P 0.00066 AFFX-BioDn-3_at 16410.8984 P 0.00020 AFFX-CreX-5_at 32975.3750 P 0.00004