where xi, xj are two expression profiles for genes i,j,k is the index of experiment (field), xik is the expression value of gene i in the experiment k.
The program clusters gene expression profiles or samples by SOM (Self-Organizing Map) algorithm.
The expression data for the set of genes is represented as a table, consisting of rows (usually corresponding to genes) and columns (or fields, usually corresponding to samples/tissues/experiments). Each row corresponds to expression measurements for the gene. Columns correspond to experiments/samples/tissues. However, this table may include not only expression data, but also other information related to genes, for example gene names, classifiers, etc. Therefore we will call the table columns as 'fields' in general case. In general, columns of the table could be of four basic types:
| IVALUE | signed integer value; | |
| FVALUE | floating point value; | |
| WORD | text without spaces inside (single word); | |
| STRING | text with spaces inside allowed. |
Basic input file format should be as follows:
; May contain comment starting from the semicolon in any line of the file NAME<tab>WORD GENEID<tab>IVALUE TISSUECANCER0<tab>FVALUE TISSUECANCER1<tab>FVALUE TISSUENORMAL0<tab>FVALUE TISSUENORMAL1<tab>FVALUE TISSUENORMAL2<tab>FVALUE #GROUP<tab>Cancer tissues TISSUECANCER0 TISSUECANCER1 #ENDGROUP #GROUP<tab>Arbitrary group TISSUECANCER1 TISSUECANCER2 TISSUENORMAL0 TISSUENORMAL1 #ENDGROUP END DATA GENE04675<tab>402<tab>6.00<tab>5.60<tab>5.97<tab>6.00<tab>6.00 GENE46890<tab>794<tab>2.77<tab>3.22<tab>5.65<tab>5.68<tab>5.68 GENE23794<tab>404<tab>5.97<tab>5.97<tab>6.00<tab>5.60<tab>5.97
In this example <tab> implies 'Tab' character symbol.
First lines (up to the "DATA" line) contain data format description. In this part of the file each line describes field description: field name and field basic type.
After the "DATA" line - data on each gene are represented. Each line correspond single cards. Field data are separated by 'tab' symbol. Double 'tab' is interpreted as missed data.
It is assumed in SetTag program that the expression data in the file are normalized and the expression levels of genes in experiments are comparable.
MolQuest version of the SelTag program can also operates with other types of files, namely, selection files. These files contain information about some selected genes or samples from the large data file in SelTag format. The selection file contain: the data file name from which selection was obtained; type of selection data (genes of samples), list of selected objects (their indices in the large data file). The selection files are in the XML format. Two examples are below.
Selection for some genes.
<?xml version="1.0" encoding="ISO-8859-5"?> <SELECTION> <HEADER name="cc_Selection5"> <DATA source="c:/data/cc.txt"/> <COMMENT><![CDATA["$F1 == "GEN14263" | $F12 >= 300"]]></COMMENT> </HEADER> <ELEMENTS type="GENES" count="9"> <![CDATA[0;1;2;10;14;15;17;26;30]]> </ELEMENTS> </SELECTION>
Selection for some fields (samples).
<?xml version="1.0" encoding="ISO-8859-5"?> <SELECTION> <HEADER name="notterman2001_set1"> <DATA source="c:/data/notterman2001_set1.txt"/> <COMMENT><![CDATA["From cc.txt data file."]]></COMMENT> </HEADER> <ELEMENTS type="FIELDS" count="10"> <![CDATA[0;1;2;3;5;6;7;18;19;30]]> </ELEMENTS> </SELECTION>
Selection files may be selected during the SelTag execution and also used by SelTag for calculation and/or visualization. Note, each selection file is linked to large data file by its name. Selection data cannot be applied to another data file.
SOM (Self-organizing map) algorithm was suggested for unsupervised learning problems solution (i.e. classification) by Kohonen [Kohonen, T. (1997) Self-Organizing Maps (Springer, Berlin)]. The algorithm provides mapping from high-dimensional data to low-dimensional space (2D). The SOM clustering was used for expression data analysis by Tamayo et al. [Tamayo P. et al (1999) Proc. Natl. Acad. Sci. USA, 96, 2907-2912]. The approach of Tomayo et al is implemented in SelTag.
An SOM has a set of nodes with a simple topology (e.g., two-dimensional grid) and a distance function d(N1,N2) on the nodes. Nodes are mapped into K-dimensional "gene expression" space (in which the i-th coordinate represents the expression level in the i-th sample, K is the number of experiments (fields)). The process of mapping is iterative (see Fig.1).
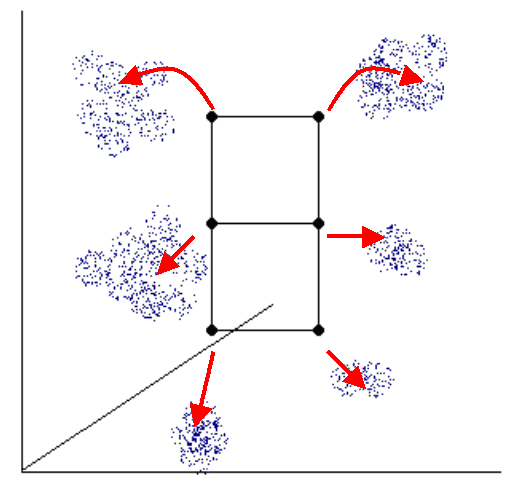
Fig. 1. The diagram shows the principle of iterative clustering of high-dimensional data points by SOM algorithm. The SOM structure is shown by black grid, data points in high-dimensional space are shown in blue. The moving of grid nodes to the regions of higher data density are shown in red.
The iterative algorithm allows moving each node to the K-dimensional space
regions with higher density of points (genes). In principle, each node will be
located near the cluster of genes in the high-dimensional space. The position
of node N at iteration i is denoted fi(N). The initial mapping
f0 is random. On subsequent iterations, a data point P is selected and
the node NP that maps nearest to P is identified. The mapping of nodes
is then adjusted by moving points toward P by the formula (Tomayo et al,1999):
fi+1(N) = fi(N) + t(d(N, NP), i) (P - fi(N)).
To perform calculation user should define the grid size (number of row and column nodes in two-dimensional grid (see Fig.1), set the maximal number of iterations and set the distance type (to calculate distance between node and data points). There are several measures of expression profile distance between two genes:
(1) Euclidean distance. This is the geometric distance in the multidimensional space.
It is computed as:
![]()
where xi, xj are two expression profiles for genes i,j,k is the index
of experiment (field), xik is the expression value of gene i in the experiment k.
(2) Squared Euclidean distance. The squared Euclidean distance can be
implemented in order to place progressively greater weight on objects that
are further apart. The squared Euclidian distance is computed as:
![]()
(see explanation above). The Euclidian and squared Euclidian distances are computed
from raw data (non-standardized), therefore they may be affected by differences in
scale among the expression values in different experiments.
(3) Manhattan distance. This distance is the average absolute difference for the set of experiments calculated by the formula
![]() .
.
In most cases, this distance measure yields results similar to the simple Euclidean distance, for this measure, the effect
of single large differences is dampened (since they are not squared).
(4) Chebychev distance. This distance is computed as dij = maxk | xik - xik |. The measure is useful when one wants to define two objects as "different" if they are different on any one of the experiments.
In SelTag all distance measures (1-3) are normalized to the number of fields involved in calculation. This is useful when take into account expression data with missing values.
Other measures involve correlation coefficient rij between two expression profiles of genes i and j.
(5) 1-rij; This measure keep close profiles with positive correlation coefficients and is useful when one wants to detect co-regulated genes.
(6) 1-|rij|; This measure keep close profiles with higher absolute value of correlation coefficients.
(7) 1+rij; This measure keep close profiles with negative value of correlation coefficients (anti-correlated).
Three types of correlation are possible for correlation distance option:
Pearson's r - Pearson's correlation coefficient.
The Pearson product moment correlation coefficient between expression profiles i and j is calculated as follows:
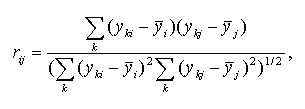
where yki is the expression level of gene i in the experiment k;
![]() is the mean
expression level of the gene i. Positive correlation implies that the
expression levels of genes i,j are related positively, the higher expression
of gene i, the higher expression of gene j. Negative correlation
means that the expression levels of genes i,j are related negatively, the
higher expression of gene i, the lower expression of gene j.
If the rij is close to zero, two expression profiles are unrelated.
is the mean
expression level of the gene i. Positive correlation implies that the
expression levels of genes i,j are related positively, the higher expression
of gene i, the higher expression of gene j. Negative correlation
means that the expression levels of genes i,j are related negatively, the
higher expression of gene i, the lower expression of gene j.
If the rij is close to zero, two expression profiles are unrelated.
Spearman r - Spearman's correlation coefficient.
This correlation coefficient is computed for ranks. Let Rki is
the rank of the expression level in the experiment k of gene i
(relatively to other experiments), Rkj is the rank of the
expression level in the experiment k of gene j.
Then Spearman's correlation coefficient is calculated by the formula
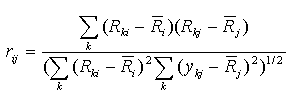
Kendall's t - Kendall's tau correlation coefficient.
To calculate Kendall's t K for data points
(yki; ykj) 2K(K - 1) pairs considered (without self-pairing, the points
in either order count as one pair).
Pairs in which yki > ymi and
ykj > ymj or
yki < ymi and ykj < ymj
are called concordant pairs (agreement between ranks), pairs with rank disagreement are called discordant pairs.
In general, t is calculated as
t = ([number of concordant] - [number of discordant]) / total number of pairs
status=done [0.0 sec] Number of gene clusters obtained 5. Cluster Sizes and Scores: Cluster 1 1 0.0000 Cluster 2 1 0.0000 Cluster 3 5 0.5954 Cluster 4 14 0.8092 Cluster 5 13 0.8647 List of selected genes, their cluster indices and scores : No DataIndex Name Cluster Score 1 1 GEN30482 1 0.0000 2 2 GEN03437 2 0.0000 3 3 GEN03687 3 0.7264 4 4 GEN24649 4 0.9291
Some lines starting from "status=" are just output the status of the calculation and can be ignored. Then the result cluster information is output: number of clusters, their list with cluster scores. Then list of selected genes with their cluster indices and scores is printed out.