Services Test Online
SNP discovery and evaluation
DESCRIPTION
SYNOPSIS
COMMANDS AND OPTIONS
CONFIGURATION FILES
LICENSE AND CITATION
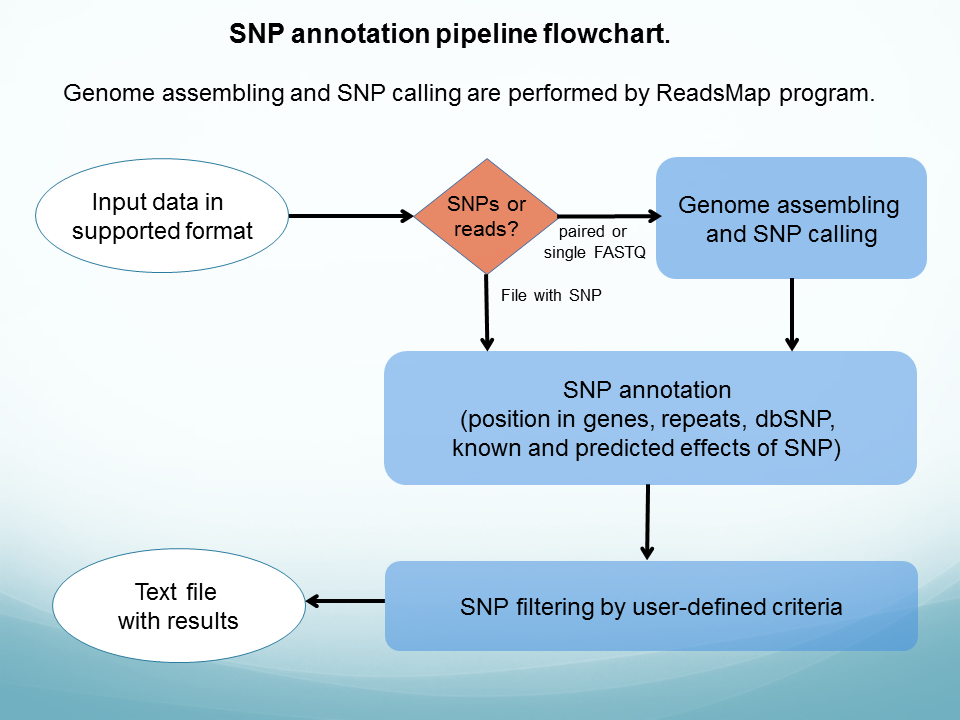
|
DESCRIPTION
SNP effect annotates a set of human SNPs. Input data contains either genome reads or a list of variations in one of supported formats. Position numbering in such a list must correspond to GRCh37/hg19 genome assembly. Variations in output file can be filtered by user-defined criteria. The program evaluates single nucleotide polymorphisms (SNPs), multiple nucleotide polymorphisms (MNPs) and insertions and deletions (indels).
The following information is reported in output file for each SNP, if applicable:
- Chromosome number, position on a chromosome, reference and observed nucleotides.
- dbSNP ID, OMIM link.
- Short description from SNPedia.
- Repetitive DNA element from UCSC database that contains an SNP.
- 5. For each gene containing a variation (human genes from UCSC database, fRNAdb database and genes which were predicted by FGENESH [1], predicted genes have IDs starting with FG_): its type (protein coding or not), database ID, orientation, position, and CDS range.
- Positions of all exons of a gene.
- Short description of a gene from UCSC database.
- Gene type: protein or RNA (not mRNA) coding.
- Position of variation in the gene: CDS or UTR, exon or intron, and whether a variation falls into a splice site, defined as two nucleotides at 5' and 3' ends of introns.
- Type of variation: synonymous, non-synonymous, stop-gain, stop-loss, insertion, deletion, multi-nucleotide, frameshift.
- Position of amino acid substitution caused by variation in translated protein.
- Total length of translated protein.
- Codon change in the gene caused by variation.
- Amino acid substitution in a protein.
- Tolerance score of a substitutions, calculated by algorithms similar with the SIFT [2] approach. The score is calculated based on occurrence of a substituted amino acid in a given position of alignment of a protein to homologs from the UniProt Knowledgebase or NCBI nr protein database. If the score is lower than a threshold (0.05), a variation is considered as damaged. Tolerance score cannot be calculated if a protein doesn’t have enough homologs.
Items 5-14 are repeated for each SNP effected gene.
References
- Solovyev V, Kosarev P, Seledsov I, Vorobyev D. Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 2006,7, Suppl 1: P. 10.1-10.12.
- Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003 Jul 1;31(13):3812-4.
SYNOPSIS
This is an example of running SNP-effect from a command line:
python SNP-effect_pipeline.py SRR701474_1.filt.fastq,SRR701474_2.filt.fastq reads_paired SRR701474.txt
COMMANDS AND OPTIONS
python SNP-effect_pipeline.py infile infile_format outfile
infile - input file name
infile_format - input file format
outfile - output file name
Supported input file formats:- vcf - VCF4.0 VCF file contains meta-information lines (start with ##), a header line (starts with #CHROM), and then data lines each containing information about position of a variation in a genome. First column is a chromosome, second is position, fourth is a reference nucleotide, fifth is a observed non-reference nucleotide or a list of such nucleotides. In the current version, meta-information and header lines are ignored.
- annovar - ANNOVAR file format. First five columns: chromosome, start position, end position, reference nucleotides and observed nucleotides. For a SNP, start and end positions are identical. Other (optional) columns are ignored.
- annovar_h - ANNOVAR file format with zygosity information (sixth column). .
- simple - simple ANNOVAR-like format most suitable for manual input of SNP data. Four columns: chromosome, position, reference nucleotide, observed nucleotide.
- 23andme - 23andMe file format. Header lines start with #. Four columns: ID, chromosome, position, observed nucleotides. References nucleotides for each variation are taken from reference genome sequence.
- cdr - VAAST file format. cdr stands for 'condenser format'. Such files are generated by VST program of VAAST package from sets of GVF files.
- reads_single - single-end reads in FASTQ format.
- reads_paired - paired-end reads in FASTQ format. Two input files must be specified, names are comma-separated without spaces.
All SNP file formats are tab-delimited, but any sequence of tabs or spaces is considered as a column separator. A reference nucleotide is taken from a reference genome sequence, so any valid DNA nucleotide (A, C, G or T) is permitted in an SNP description string. All positions must correspond to GRCh37/hg19 genome assembly, and base numbering starts from 1.
Input data examples:
VCF:
1 10469 . C G 100 PASS . GT:AP 1|0:0.740,0.450 1 1477244 rs7290 T C 100 PASS . GT:AP 1|1:0.700,0.820 2 183699584 rs7775 G C 100 PASS . GT:AP 0|1:0.440,0.695
ANNOVAR:
1 10469 10469 C G 1 1477244 1477244 T C comments: rs7290 2 183699584 183699584 G C
Simple:
1 10469 C G 1 1477244 T C 2 183699584 G C
23andme:
. 1 10469 CG rs7290 1 1477244 CT rs7775 2 183699584 CCExample of output file:
#VARIATION chr1 10469 C --> G INTERGENIC Name: between FR137075 and uc010nxq.1 #VARIATION chr1 1477244 T --> C dbSNP ID: rs7290 #INTERSECTED GENE Name: uc001agd.3 Strand: - Region: 1477053..1510262 CDS: 1477446..1509937 Exons: 1477053..1477547 1479249..1479367 1480243..1480382 1500153..1500296 1509858..1510262 Description: Homo sapiens SSU72 RNA polymerase II CTD phosphatase homolog (S. cerevisiae) (SSU72), mRNA. Type of gene: Protein coding Clinical significance: unknown Variation location: Out of CDS, 3' UTR. #VARIATION chr2 183699584 G --> C FREQ: 0.669682 dbSNP ID: rs7775 OMIM link: http://omim.org/entry/605083#0001#INTERSECTED GENE Name: uc002upa.2 Strand: - Region: 183698005..183731498 CDS: 183699576..183731280 Exons: 183698005..183699692 183702676..183702739 183703137..183703341 183707206..183707271 183723514..183723561 183730803..183731498 Description: Homo sapiens frizzled-related protein (FRZB), mRNA. Type of gene: Protein coding Clinical significance: osteoarthritis; colorectal cancer; Defects in FRZB are associated with susceptibility to osteoarthritis type 1(OS1); Variation location: Exon 6 Position in protein: 324 Protein length: 325 Codon: CGC => GGC Translation: R => G Tolerance Score: 0.01 (damaging)
CONFIGURATION FILES
Comfiguration file name is snp.ini.
Path to files with data are set in snp.ini in [global] section. Filtering parameters are set in [sift_score_filter] and [add_filters] sections.
enabled=0 turns on SIFT score filter.
show_only_scored=yes means ignoring variations without SIFT scores.
If compare_less=0, SNPs with scores more than compare_level are printed in output file, compare_less=1 outputs SNPs with scores less than compare_level. Small values of SIFT score correspond to variations that are not dmaged. Usual cutoff level (compare_level) is 0.05.
With coding_region only SNPs in coding region of genes are printed in output file.
omim and snpedia options are for filtering only SNPs with information in OMIM database or SNPedia.
[global] base_dir = ./data pssm_dir = pssm snp_dir = dbSNP omim_dir = omim seq_dir = ref_fa annot_fname = knownGene.fg2 gaps_fname =gaps.kg2 repeats_fname =norepeats ;hg19_rmsk.txt info_dir = annotations info_fname = knownGene.ann8.chr chr_list = 1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,X,Y ;comma separated snpedia_fname=snpedia/snpedia.txt gad_fname=gad/GAD.txt freq_dir = frequencies tpl_dir = ../templates readsMap_dir = ./readsMap [sift_score_filter] enabled=1 show_only_scored=yes compare_less=0 compare_level=0.05 [add_filters] coding_region=1 omim=0 snpedia=0 snpedia=0
LICENSE AND CITATION
Copyright © Softberry, Inc., 2014-2016