Services Test Online
Adapter_Trim
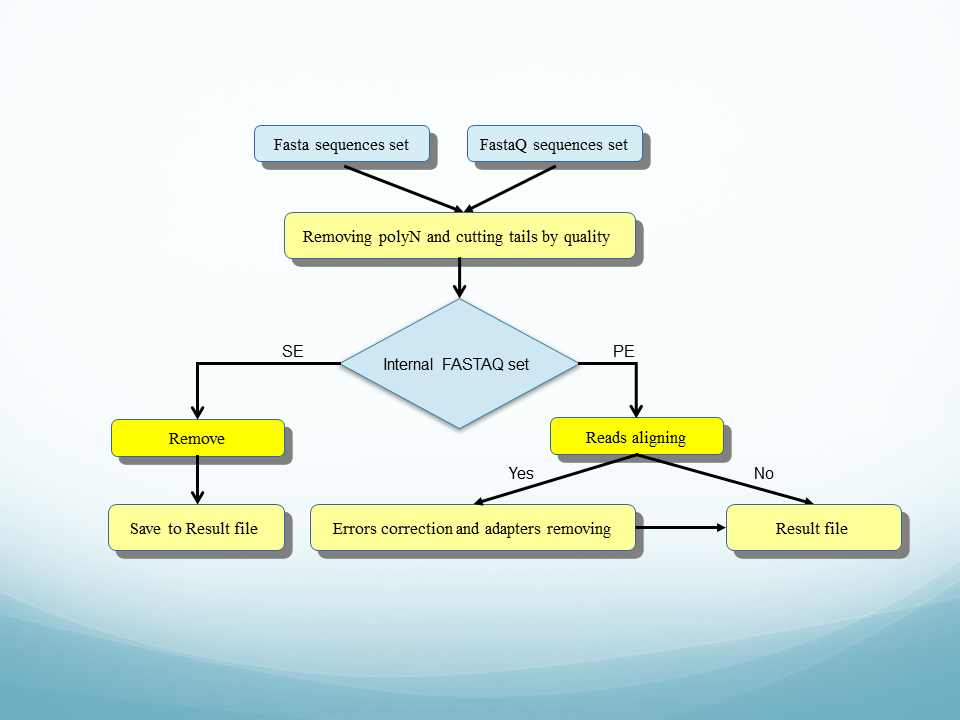
CONTENTS
Synopsis
Description
Build
Commands And Options
Sample
Accuracy
License And Citation
Downloads
SYNOPSIS
Try to determinate adapters sequences in FASTQ pair-ends reads. Result will be printed in stdout.
./adapter_trim SRR330569_1.fastq SRR330569_2.fastq -ifastq - phread33 -PE -o:adapter_trim.cfg -analyze -j:8
Scan FASTQ pair-ends reads and remove adapters. Source reads are in two files. Result will be saved to one file in FASTA format.
./adapter_trim SRR519624_1.fastq SRR519624_2.fastq -ifastq -phread33 -PE -o:adapter_trim.cfg -adapters_trim -adpt1_seq:AGATCGGAAGAGCGGTTCAGCAGGAATGCCGAGACCGATCTCGTATGCCGTCTTCTGCTTG -adpt2_seq:AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATT -min_read_flen:0 -min_read_slen:0 -to_one_file -to_fasta -j:7
Remove polyN and low quality tails from reads in FASTQ format. Result will be saved to two files in FASTQ format.
./adapter_trim SRR519624_1.fastq SRR519624_2.fastq -ifastq - phread33 -PE -o:adapter_trim.cfg -cut_polyN -cut_qual -cut_qual_level:15 -to_fastq -to_two_files
DESCRIPTION
adapter_trim is a newest program for the preparation of short sequences (reads) sets for further analysis. The adapter_trim main goal is removing adapters from reads, but you can use it for the task of searching and removing polyN tails and cutting sequence by quality.
BUILD
Run the *_buid_all.sh script in the ./build folder
Assembled programs are to be placed to the ./bin folder
COMMANDS AND OPTIONS
Define adapters sequences . More than one adapter sequences can be use for each set of reads.
-adpt1_seq:AGATC...GA Read1 adapter sequence, may be set more than once -adpt2_seq:GGACC...TA Read2 adapter sequence, may be set more than once
Input files format. .
You can specify only one file for single ends reads. There are two valid variants for "MP" and "PE" reads:
1. Reads are separated in two files. In this case first read from pair must be placed in one file
and second read in other file. Obviously the sequence number of reads from one pair must be equal.
2. All reads in one file. In this case sequences of paired reads are supposed to be in order (even/odd).
-PE Input file(s) is the "PE" reads (default).
-MP Input file(s) is the "MP" reads.
-SE Input file is the "SE" (Single ends) reads.
Input files parameters
-fasta input file(s) are in FASTA format.(Default)
-fastq input file(s) are in FASTQ format.
You are need to specify FASTQ version by one of this options:
-phread33 Sanger and Illumina 1.8+ version format
-phread64 Illumina 1.3+ and Illumina 1.5+ version format
For FASTA format each nucleotide have fixed quality. You can change this value by -def_fasta_qua: option.
-def_fasta_qua:XX Default quality for input file(s) in FASTA format. Numeric value (Default is 20)
Select mode . You must set at least one mode in command line, otherwise no any actions will be done,
-adapters_trim Adapter trimmer mode. Search and remove adapters.
-cut_polyN Cut plyN mode. Search and remove polyN tails.
-cut_qual Cut by quality mode. Search and remove tails with bad quality. Use it with -cut_qual_level: option.
-cut_qual_level:XX Numerical value (0-40) for cut by quality threshold. (Default is 0)
Multiprocessing .
-j:XX Number of processes for multiprocessing regime.
Output files parameters.
-no_save_name Skip name of read. Name will be replaced by number. C
-to_fasta Save result in FASTA format.
-to_fastq Save result in FASTQ format.
-to_one_file Save result reads pairs in single file. Read1 will be odd and read2 will be even.
-to_two_files Save result reads pairs in differ file. So one file from pair will be saved in file with suffix ".1" and other in file with suffix ".2". Single reads will be saved in file with suffix ".0".
-no_qual_limiter Allow store quality values larger than 40.;
-join Join overlapped paired reads in one sequence.
adaper_trim can sort source base to multiply files. You can regulate this behavior by next options:
-min_read_flen:XXX Minimal length of long reads. Reads shorter than XXX will be placed in base of short reads. Default is 55.
-min_read_slen:XXX Minimal length of short reads. Reads shorter than XXX will be skipped. Default 15.
Set both of this in 0 and all result will be placed in one or two (according -to_two_files or -to_one_file options) file(s).
Result destination
-ad_trim_path:path Destination folder.
-ad_base_name:name Prefix for output files. If it set the basename will be replaced by given name.
Advanced options
-analyze Try to analyze source set of reads and offer the most probable variants of adapters. It`s strongly recommended to repeat analyze procedure after first run, with using the sequences which were found after the first run as seed (see the sample).
-adapter_len_max:XX Use not greater then XX nucleotides from given adapters.
-store_quality For FASTA output. Save quality string in FASTA name.
-ad_max_pass:XX While align - gaps length sum in adapter must be less then XX.
-read_max_pass:XX While align - gaps length sum in read must be less then XX.
-max_gap_len:XX Maximum single gap length must be less then XX.
-cut_agressivity:XX Aggressivity level for all types of cutting.(1...inf, default 2.2).
-cut_hole:XX Length of "bad" regions for cutting.
-mp_cross_only Process only overlapped MP reads.
-adpt1_shift:val Shift adapter in read1. Use 0 (default) if unsure.
-adpt2_shift:val Shift adapter in read2. Use 0 (default) if unsure.
SAMPLE
First run – search a seed.
> ./adapter_trim SRR1611127_1.fastq SRR1611127_2.fastq -ifastq -phread33 -PE -o:adapter_trim.cfg -analyze -j:8
Resolve Default adapter1
Resolve Default adapter2
First File.
Second File.
Done... Execution time: 1.638169644 sec.
---------------- Read 1 Adapter. -----------------------------------
>Read 1 Adapter.
AGATCGGAAGAGCACACGTCTGAACTC
It may be one of...
>Illumina Multiplexing PCR Primer 2.01
AGATCGGAAGAGCACACGTCTGAACTC CAGTCAC
>Illumina Multiplexing Index Sequencing Primer
AGATCGGAAGAGCACACGTCTGAACTC CAGTCAC
>Illumina Multiplexing Read2 Sequencing Primer
AGATCGGAAGAGCACACGTCTGAACTC CAGTCAC
...
---------------- Read 2 Adapter. -----------------------------------
>Read 2 Adapter.
AGATCGGAAGAGCGTCGTGTAGGGAAAGA
It may be one of...
>TruSeq Universal Adapter
AGATCGGAAGAGCGTCGTGTAGGGAAAGA GTGTAGATCTCGGTGGTCGCCGTATCATT
>Illumina Single End PCR Primer 1
AGATCGGAAGAGCGTCGTGTAGGGAAAGA GTGTAGATCTCGGTGGTCGCCGTATCATT
...
Second run – analyze with the seed using.
> ./adapter_trim SRR1611127_1.fastq SRR1611127_2.fastq -ifastq -phread33 -PE -o:adapter_trim.cfg -analyze -j:8 -adpt1_seq:AGATCGGAAGAGCACACGTCTGAACTC -adpt2_seq:AGATCGGAAGAGCGTCGTGTAGGGAAAGA Resolve user defined adapter1 Resolve user defined adapter2 First File. Second File. Done... Execution time: 1.848383758 sec. ---------------- Read 1 Adapter. ----------------------------------- >Read 1 Adapter. AGATCGGAAGAGCACACGTCTGAACTCCAGTCACATTCACTGATCTCGTATGCCGTCTTCTGCTTGAA It may be one of... >TruSeq Adapter, ATTCACTG AGATCGGAAGAGCACACGTCTGAACTCCAGTCACATTCACTGATCTCGTATGCCGTCTTCTGCTTG ---------------- Read 2 Adapter. ----------------------------------- >Read 2 Adapter. AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATTAAAAAAAAAA It may be one of... >TruSeq Universal Adapter AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATT >Illumina Single End PCR Primer 1 AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATT >Illumina Paried End PCR Primer 1 AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATT >Illumina Multiplexing PCR Primer 1.01 AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATT
Trimming
> ./adapter_trim SRR1611127_1.fastq SRR1611127_2.fastq -ifastq -phread33 -PE -o:adapter_trim.cfg -adapters_trim -adpt1_seq:AGATCGGAAGAGCACACGTCTGAACTCCAGTCACATTCACTGATCTCGTATGCCGTCTTCTGCTTG -adpt2_seq:AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATT -min_read_flen:0 -min_read_slen:0 -to_one_file -to_fasta -j:8
TRIMMING ACCURACY
We try to compare results of adapter trimming by adapter_trim and by skewer (http://sourceforge.net/projects/skewer/files/) programs with using the same adapter sequences. For the source data the reads of Arabidopsis thaliana was used. The results of adapter trimming was aligned to full genome. Quality indicators of alignment were used as a quality measure for trimming results.
Skewer
Total reads 34683594.command line: ./skewer-0.1.123-linux-x86_64 -l 0 -r 0.3 -x adapter1.fa -y adapter2.fa SRR519624_1.fastq SRR519624_2.fastq
Reads aligned with homology > 95% 27122070 (78.199%)
Total nucleotides 3462985140.
Summary alignment length 2670451726.
Matched nucleotides 2622437780 (75.728%)
adapter_trim
Total reads 34683594command line: ./adapter_trim SRR519624_1.fastq SRR519624_2.fastq -PE -ifastq -phread33 -o:adapter_trim.cfg -adpt1_seq:AGATCGGAAGAGCGGTTCAGCAGGAATGCCGAGACCGATCTCGTATGCCGTCTTCTGCTTG -adpt2_seq:AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATT -min_read_flen:0 -min_read_slen:0 -to_one_file -to_fasta -store_quality -j:7 -adapters_trim
Reads aligned with homology > 95% 27149366 (78.277%)
Total nucleotides 3461498047
Summary alignment length 2707527341
Matched nucleotides 2640217552 (76.274%)
Thus it can be seen that the adapter_trim performs better clean quality.
LICENSE AND CITATION
adapter_trim is a free for academic usage. Please contact to softberry@softberry.com in otherwise.