Services Test Online
FGENESB_annotator is a package for automatic annotation of bacterial genomes and bacterial community sequences annotation developed in Softberry Inc. by Victor Solovyev and Asaf Salamov (modified by Peter Kosarev)
This manual includes:
- a) Introduction/Description
- b) Explanation of Fgenesb_annotator output
- c) How to run scripts of Fgenesb_annotator; configuration (paths) file
- d) Installation
- e) Conversion to GenBank format
- f) Conversion to Sequin format
- g) Appendix. Running gene/operon prediction steps in manual mode. Actual scripts.
a) Introduction/Description
To identify protein and RNA genes in bacterial genomic sequences or environmental samples, Softberry developed Fgenesb_annotator pipeline that provides completely automatic, comprehensive annotation of bacterial sequences. The pipeline includes protein, tRNA and rRNA genes identification, finds potential promoters, terminators and operon units.
Predicted genes are annotated based on comparison with known proteins. The package provides options to work with a set of sequences such as scaffolds of bacterial genomes or short reads of DNA extracted from a bacterial community. The final annotation can be presented in GenBank form to be readable by visualization software such as Artemis [1] and Genome Explorer (fig. 1 and 2). The gene prediction algorithm is based on Markov chain models of coding regions and translation and termination sites. For annotation of mixed bacterial community, we use special parameters of gene prediction computed based on a large set of known bacterial sequences. Operon models are based on distances between ORFs, frequencies of different genes neighboring each other in known bacterial genomes, and information from predicted potential promoters and terminators. The parameters of gene prediction are automatically trained during initial steps of sequence analysis, so the only input necessary for annotation of a new genome is its sequence. Optionally, parameters from closely related genomes can be used, instead of training new parameters. Bacterial gene/operon prediction and annotation requires, besides Fgenesb_annotator programs and scripts, BLAST, NCBI Non-Redundant database (NR), and a file reconstructed from COG database [2]. RRNA genes are annotated using BLAST similarity with all known bacterial rRNAs database. For prediction of tRNA genes, the pipeline uses tRNAscan-SE package [3].
1. K. Rutherford, J. Parkhill, J. Crook, T. Horsnell, P. Rice, M-A. Rajandream and B. Barrell (2000) Artemis: sequence visualisation and annotation. Bioinformatics 16 (10) 944-945.
2. Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. (2001) The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 29, 22-28.
3. Lowe, T.M. & Eddy, S.R. (1997) "tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence", Nucl. Acids Res., 25, 955-964.
- The main features of Fgenesb_annotator are:
- Automatic training of gene finding parameters for new bacterial genomes using only genomic DNA as an input
- Optionally, pre-learned parameters from related organism can be used
- Optionally, generic Bacterial, Archaebacterial, or combined parameters can be used
- Mapping of tRNA and rRNA genes
- Mapping of bacterial ribosomal proteins from DB and selecting good mappings
- Highly accurate Markov chains-based gene prediction
- Prediction of promoters and terminators
- Operon prediction based on distances between ORFs and frequencies of different genes neighboring each other in known bacterial genomes, as well as on promoter and terminator predictions
- Automatic annotation of predicted genes by homology with protein databases such as COG, KEGG, NR and/or custom protein databases.
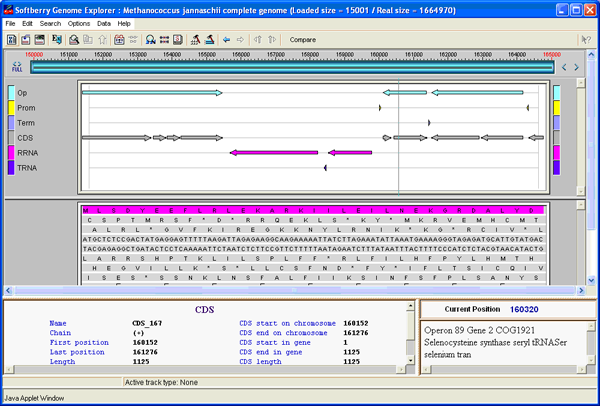
The package includes options to work with a set of sequences such as scaffolds of bacterial genomes, or short sequencing reads extracted from bacterial communities. For community sequence annotation, we developed ABsplit program that separates archaebacterial and eubacterial sequences (available separately). Final annotation can be presented in GenBank format to be readable by visualization software such as Artemis or Softberry Bacterial Genome Explorer (fig. 1 and 2, GenBank parser is available separately).
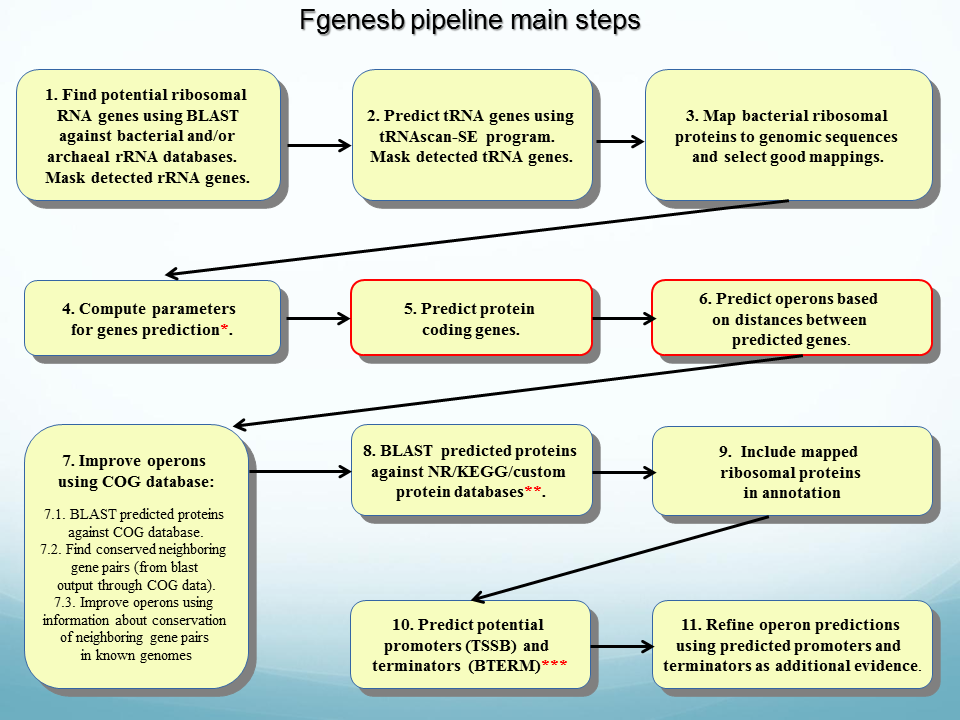
Main Steps of FGENESB annotation.
Many steps are optional and can be switched ON/OFF in configuration file.
STEP 1. Finds all potential ribosomal RNA genes using BLAST against bacterial and/or archaeal rRNA databases, and masks detected rRNA genes.
STEP 2. Predicts tRNA genes using
tRNAscan-SE program (Washington University) and masks detected tRNA genes.
STEP 3. Mapping bacterial ribosomal proteins to genomic sequences and selecting good mappings.
STEP 4. Initial predictions of long ORFs that are used as a starting point for calculating parameters for gene prediction. Iterates until stabilizes. Generates parameters such as 5th-order in-frame Markov chains for coding regions, 2nd-order Markov models for region around start codon and upstream RBS site, stop codon and probability distributions of ORF lengths.
STEP 5. Predicts operons based only on distances between predicted genes.
STEP 6. Runs BLAST for predicted proteins against COG database, cog.pro
STEP 7. Finds conserved operonic pairs from blast output through cog data.
STEP 8. Uses information about conservation of neighboring gene pairs in known genomes to improve operon prediction.
STEP 9. Runs BLAST for predicted proteins against protein databases such as KEGG, NR and/or custom protein databases.
STEP 10. Adds names of homologs from COG/KEGG/NR/etc. (found through BLAST) to annotation file (file with prediction results).
STEP 11. Predicts potential promoters (TSSB1 program) or terminators (BTERM) in upstream and downstream regions, correspondingly, of predicted genes. BTERM is the program predicting bacterial-independent terminators with energy scoring based on discriminant function of hairpin elements.
STEP 12. Refines operon predictions using predicted promoters and terminators as additional evidences.
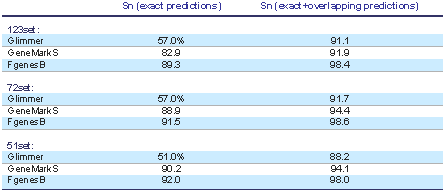
FGENESB gene prediction engine is one of the most accurate prokaryotic gene finders available: see Table 1 for its comparison with two other popular gene prediction programs.
Table 1. Comparison of three popular bacterial gene finders. Accuracy estimate was done on a set of difficult short genes that was previously used for evaluating other bacterial gene finders (http://opal.biology.gatech.edu/GeneMark/genemarks.cgi). First set (51set) has 51 genes with at least 10 strong similarities to known proteins. Then 72set has 72 genes with at least two strong similarities, and 123set has 123 genes with at least one protein homolog.
Here are the prediction results on these three sets for GeneMarkS and Glimmer (calculated by Besemer et al. (2001) Nucl. Acids Res. 29:2607-2618) and FGENESB gene prediction engine (calculated by Softberry).
All prediction components of FGENESB are extremely fast (minutes per genome). The limiting stage is BLAST annotation, which for E.coli genome takes around 12 hours on a single processor. Using multiple processors and corresponding BLAST would speed up annotation proportionally.
b) Explanation of Fgenesb_annotator output
Example of FGENESB output:
Prediction of potential genes in microbial genomes
Time: Tue Aug 22 11:21:15 2006
Seq name: gi|15807672|ref|NC_001264.1| Deinococcus radiodurans R1 (partial sequence)
Length of sequence - 54865 bp
Number of predicted genes - 48, with homology - 48
Number of transcription units - 18, operons - 13 average op.length - 3.3
N Tu/Op Conserved S Start End Score
pairs(N/Pv)
- TRNA 147 - 222 78.9 # Arg CCG 0 0
+ TRNA 315 - 398 63.6 # Leu TAG 0 0
+ 5S_RRNA 521 - 637 100.0 # AB001721 [D:2735..2851]
+ SSU_RRNA 698 - 2181 100.0 # SSU_RRNA ##
+ LSU_RRNA 2302 - 5345 100.0 # BX248583 [R:613128..616171]
+ Prom 5304 - 5363 41.4
1 1 Op 1 22/0.000 + CDS 5410 - 6300 498 ## COG1192 ATPases involved ...
2 1 Op 2 . + CDS 6297 - 7178 502 ## COG1475 Predicted ...
+ Term 7203 - 7253 9.1
- Term 7191 - 7241 14.2
3 2 Tu 1 . - CDS 7283 - 8746 909 ## COG1012 NAD-dependent ...
- Prom 8792 - 8851 2.8
4 3 Tu 1 . + CDS 8802 - 9533 302 ## COG2068 Uncharacterized ...
+ Term 9779 - 9818 3.8
- Term 9527 - 9567 9.0
5 4 Op 1 2/0.125 - CDS 9584 - 10762 1005 ## COG1063 Threonine ...
6 4 Op 2 . - CDS 10759 - 11457 666 ## COG5637 Predicted integral ...
- Prom 11697 - 11756 2.4
7 5 Op 1 37/0.000 + CDS 11704 - 12609 872 ## COG1131 ABC-type multidrug ...
8 5 Op 2 5/0.000 + CDS 12726 - 13517 812 ## COG0842 ABC-type multidrug ...
9 5 Op 3 15/0.000 + CDS 13674 - 14684 1028 ## COG4585 Signal transduction ...
10 5 Op 4 . + CDS 14681 - 15316 506 ## COG2197 Response regulator ...
...
47 18 Op 1 . - CDS 53783 - 54703 431 ## DRA0045 hypothetical ...
48 18 Op 2 . - CDS 54700 - 54864 91 ## DRA0046 hypothetical ...
Predicted protein(s)
>gi|15807672|ref|NC_001264.1| GENE 1 5410 - 6300 498 296 aa, chain + ## HITS:3
COG:DRA0001 KEGG:FRAAL2247 NR:6460595 ## COG: DRA0001 COG1192 #
Protein_GI_number: 15807673 # Func_class: D Cell cycle control, cell division, chromosome
partitioning # Function: ATPases involved in chromosome partitioning # Organism: Deinococcus
radiodurans # 37 296 1 260 260 459 100.0 1e-129 ## KEGG: FRAAL2247 #
Name: not_defined # Def: chromosome partitioning protein (partial match) [EC:2.7.10.2] #
Organism: F.alni # Pathway: not_defined # 48 283 50 291 302 118 35.0 5e-26 ##
NR: gi|6460595|gb|AAF12301.1| chromosome partitioning ATPase, putative, ParA family
[Deinococcus radiodurans R1]^Agi|15807673|ref|NP_285325.1| chromosome partitioning ATPase, putative,
ParA family [Deinococcus radiodurans R1] # 37 296 1 260 260 459 100.0 1e-128
VLKNHLFLRNLIFSVLPVVQHFLTFKEEQSIADLSDMVSAVKTLTVFNHAGGAGKTSLTL
NVGYELARGGLRVLLLDLDPQANLTGWLGISGVTREMTVYPVAVDGQPLPSPVKAFGLDV
IPAHVSLAVAEGQMMGRVGAQGRLRRALAEVSGDYDVALIDSPPSLGQLAILAALAADQM
IVPVPTRQKGLDALPGLQGALTEYREVRPDLTVALYVPTFYDARRRHDQEVLADLKAHLS
PLARPVPQREAVWLDSTAQGAPVSEYAPGTPVHADVQRLTADIAAAIGVAYPGENA
>gi|15807672|ref|NC_001264.1| GENE 2 6297 - 7178 502 293 aa, chain + ## HITS:3
COG:DRA0002 KEGG:SAR11_0354 NR:12230476 ## COG: DRA0002 COG1475 #
Protein_GI_number: 15807674 # Func_class: K Transcription # Function: Predicted transcriptional
regulators # Organism: Deinococcus radiodurans # 1 293 1 293 293 478 100.0
1e-135 ## KEGG: SAR11_0354 # Name: parB # Def: chromosome partitioning protein [EC:2.7.7.-] #
Organism: P.ubique # Pathway: not_defined # 10 200 12 177 282 107 36.0 7e-23 ##
NR: gi|12230476|sp|Q9RZE7|PARB2_DEIRA Probable chromosome 2 partitioning protein parB
(Probable chromosome II partitioning protein parB)^Agi|6460594|gb|AAF12300.1| chromosome partitioning
protein, ParB family [Deinococcus radiodurans R1]^Agi|15807674|ref|NP_285326.1| chromosome partitioning
protein, ParB family [Deinococcus radiodurans R1] # 1 293 1 293 293 478 100.0
1e-133
MTRRRPERRRDLLGLLGETPVDLSQANDIRALPVNELKVGSTQPRRSFDLERLSELAESI
RAHGVLQPLLVRSVDGQYEIVAGERRWRAAQLAGLAEVPVVVRQLSNEQARAAALIENLQ
RDNLNVIDEVDGKLELIALTLGLEREEARKRLMQLLRAVPGDEHEQLDQVFRSMGETWRT
FAKNKLRILNWPQPVLEALRAGLPLTLGSVVASAPPERQAELLKLAQNGASRSQLLQALQ
TPSQTSAVTPEHFAKVLSSKRFLSGLDTPTREALDRWLARMPERVRQAIDEQS
...
Example of FGENESB output in GenBank format (scripts run_tgb.pl, togenbank.pl):
gene complement(147..222)
/gene="Arg CCG"
tRNA complement(147..222)
/gene="Arg CCG"
/product="tRNA-Arg"
/note="Arg CCG 0 0"
gene 315..398
/gene="Leu TAG"
tRNA 315..398
/gene="Leu TAG"
/product="tRNA-Leu"
/note="Leu TAG 0 0"
gene 521..637
/gene="AB001721 [D:2735..2851]"
rRNA 521..637
/gene="AB001721 [D:2735..2851]"
/product="5S ribosomal RNA"
/note="AB001721 [D:2735..2851]"
gene 698..2181
/gene="SSU_RRNA"
rRNA 698..2181
/gene="SSU_RRNA"
/product="16S ribosomal RNA"
/note="SSU_RRNA"
gene 2302..5345
/gene="BX248583 [R:613128..616171]"
rRNA 2302..5345
/gene="BX248583 [R:613128..616171]"
/product="23S ribosomal RNA"
/note="BX248583 [R:613128..616171]"
promoter 5304..5363
CDS 5410..6300
/function="ATPases involved in chromosome partitioning"
/note="Operon 1 Gene 1 COG1192 ATPases involved in
chromosome partitioning"
/translation="VLKNHLFLRNLIFSVLPVVQHFLTFKEEQSIADLSDMVSAVKTL
TVFNHAGGAGKTSLTLNVGYELARGGLRVLLLDLDPQANLTGWLGISGVTREMTVYPV
AVDGQPLPSPVKAFGLDVIPAHVSLAVAEGQMMGRVGAQGRLRRALAEVSGDYDVALI
DSPPSLGQLAILAALAADQMIVPVPTRQKGLDALPGLQGALTEYREVRPDLTVALYVP
TFYDARRRHDQEVLADLKAHLSPLARPVPQREAVWLDSTAQGAPVSEYAPGTPVHADV
QRLTADIAAAIGVAYPGENA"
/transl_table=11
CDS 6297..7178
/function="Predicted transcriptional regulators"
/note="Operon 1 Gene 2 COG1475 Predicted transcriptional
regulators"
/translation="MTRRRPERRRDLLGLLGETPVDLSQANDIRALPVNELKVGSTQP
RRSFDLERLSELAESIRAHGVLQPLLVRSVDGQYEIVAGERRWRAAQLAGLAEVPVVV
RQLSNEQARAAALIENLQRDNLNVIDEVDGKLELIALTLGLEREEARKRLMQLLRAVP
GDEHEQLDQVFRSMGETWRTFAKNKLRILNWPQPVLEALRAGLPLTLGSVVASAPPER
QAELLKLAQNGASRSQLLQALQTPSQTSAVTPEHFAKVLSSKRFLSGLDTPTREALDR
WLARMPERVRQAIDEQS"
/transl_table=11
terminator 7203..7253
terminator complement(7191..7241)
CDS complement(7283..8746)
/function="NAD-dependent aldehyde dehydrogenases"
/note="Operon 2 Gene 1 COG1012 NAD-dependent aldehyde
dehydrogenases"
/translation="MTTTDLRTTYSSVTRSQAYFDGEWRNAPRNFEVRHPGNGEVIGE
VADCTPTDARQAIDAAEVALREWRQVNPYERGKILRRWHDLMFEHKEELAQLMTLEMG
KPISETRGEVHYAASFIEWCAEEAGRIAGERINLRFPHKRGLTISEPVGIVYAVTPWN
FPAGMITRKAAPALAAGCVMILKPAELSPMTALYLTELWLKAGGPANTFQVLPTNDAS
ALTQPFMNDSRVRKLTFTGSTEVGRLLYQQAAGTIKRVSLELGGHAPFLVFDDADLER
AASEVVASKFRNSGQTCVCTNRVYVQRGVAEEFIRLLTEKTAALQLGDPFDEATQVGP
VVEQAGLDKVQRQVQDALTKGAQATTGGQVSSGLFFQPTVLVDVAPDSLILREETFGP
VAPVTIFDTEEEGLRLANDSEYGLAAYAYTRDLGRAFRIAEGLEYGIVGINDGLPSSA
APHVPFGGMKNSGVGREGGHWGLEEYLETKFVSLGLS"
/transl_table=11
promoter complement(8792..8851)
...
BASE COUNT 11009 a 16099 c 16880 g 10877 t
ORIGIN
1 tctttgctcg ccatacccaa agtctacacg ctgattttca cgtttccaga ccctgccctc
61 tcgctactca gctctccaag tttgctcgct tgatgaatga tcaaatcttt taaagataaa
121 agccatgcgt gaggctagat caacccttgt gcccccggca ggattcgaac ctgcggcctt
...
54841 gtcgcccagt tgaatggctc gccac
//
Example of FGENESB output in Sequin format:
>Feature test_seq
222 147 gene
locus_tag C8J_0001
222 147 tRNA
product tRNA-Arg
inference profile:tRNAscan-SE:1.23
315 398 gene
locus_tag C8J_0002
315 398 tRNA
product tRNA-Leu
inference profile:tRNAscan-SE:1.23
521 637 gene
locus_tag C8J_0003
521 637 rRNA
product 5S ribosomal RNA
698 2181 gene
locus_tag C8J_0004
698 2181 rRNA
product 16S ribosomal RNA
2302 5345 gene
locus_tag C8J_0005
2302 5345 rRNA
product 23S ribosomal RNA
5304 6300 gene
locus_tag C8J_0006
5304 5363 promoter
5410 6300 CDS
product hypothetical protein
note similar to D.radiodurans chromosome partitioning ATPase ...
protein_id gnl|bbsrc|C8J_0006
inference ab initio prediction:Fgenesb:2.0
6297 7253 gene
locus_tag C8J_0007
6297 7178 CDS
product chromosome partitioning protein, ParB family
protein_id gnl|bbsrc|C8J_0007
inference ab initio prediction:Fgenesb:2.0
7203 7253 terminator
8851 7191 gene
locus_tag C8J_0008
7241 7191 terminator
8746 7283 CDS
product succinate-semialdehyde dehydrogenase
EC_number 1.2.1.16
protein_id gnl|bbsrc|C8J_0008
inference ab initio prediction:Fgenesb:2.0
8851 8792 promoter
...
Description of Fgenesb_annotator output fields:
For each genomic sequence (complete genome, scaffold, read, etc.) the program lists locations of predicted ORFs, rRNAs, tRNAs, promoters and terminators.
ORFs are labeled as CDS and provided with their order number in a sequence and an indicator of whether they are transcribed as a single transcription unit (Tu) or in operons (Op) (of course these are predictions).
If an ORF has a homolog, its short name is provided after a "##" separator (here name of only
one homolog - either from COG, KEGG, or NR - is given; best homologs from all databases are listed in ID lines of predicted proteins, see below).
For example:
5 4 Op 2 + CDS 2737 - 3744 871 ## COG0673 Predicted dehydrogenases
is description for predicted gene number 5 in 4th Operon with coordinates 2737 - 3744 in the '+' strand and it is the second gene in operon.
Coding chain for this CDS (+) means a direct chain, (-) means a complementary chain.
871 is a score of gene homology assigned by BLAST.
COG0673 is an ID of its homolog from the COG database.
In other words, first column lists an ordered number of predicted CDS, starting from beginning of a sequence; second column - number of predicted operon/TU, and fourth column - number of gene in an operon (always 1 for a TU).
For some operons, we report supportive evidence related to conservation in relative locations of genes in predicted operon in different bacteria. For example:
3 2 Op 1 4/0.002 + CDS 3193 - 3405 278 ## COG2501 Uncharacterized ACR
Here, in 4/0.002, 4 is a number of observations of this gene being next to one of its neighbors on known bacterial genomes (we call it N-value), while 0.002 is a P-value, an empirical probability of observing N occurrences of genes being adjacent by random chance. P is a very approximate measure. For all P<0.0001, the value in output is 0.000.
At the end of annotation, we also provide protein products of predicted genes in fasta format, with full name of homolog and homology scores according to BLAST.
Information about homologs is given in ID lines of predicted proteins, for example:
>gi|15807672|ref|NC_001264.1| GENE 7 11704 - 12609 872 301 aa, chain + ## HITS:3 COG:DRA0007 KEGG:DRA0007 NR:6460585 ## COG: DRA0007 COG1131 # Protein_GI_number: 15807679 # Func_class: V Defense mechanisms # Function: ABC-type multidrug transport system, ATPase component # Organism: Deinococcus radiodurans # 1 301 1 301 301 503 100.0 1e-142 ## KEGG: DRA0007 # Name: not_defined # Def: putative ABC-2 type transport system ATP-binding protein # Organism: D.radiodurans # Pathway: ABC transporters - General [PATH:dra02010] # 1 301 1 301 301 503 100.0 1e-142 ## NR: gi|6460585|gb|AAF12291.1| ABC transporter, ATP-binding protein, putative [Deinococcus radiodurans R1]^Agi|15807679|ref|NP_285331.1| ABC transporter, ATP-binding protein, putative [Deinococcus radiodurans R1] # 1 301 1 301 301 503 100.0 1e-141 MITTFEQVSKTYGHVTALSDFNLTLRTGELTALLGPNGAGKSTAIGLLLGLSAPSAGQVR VLGADPRRNDVRARIGAMPQESALPAGLTVREAVTLFASFYPAPLGVDEALALADLGPVA GRRAAQLSGGQKRRLAFALAVVGDPELLLIDEPTTGMDAQSRAAFWEAVTGLRARGRTIL LTTHYLEEAERTADRVVVMNGGRILADDTPQGLRSGVGGARVSFVSDLVQAELERLPGVS AVQVDAAGRADLRTSVPEALLAALIGSGTTFSDLEVRRATLEEAYLQLTGPQDMTAVTRS A
While looking a bit complex for a human eye, it is well suited for parsing by a program.
ID lines of predicted proteins consist of the following parts that are separated from each
other by "##" separator:
>gi|15807672|ref|NC_001264.1| GENE 7 11704 - 12609 872 301 aa, chain +
(sequence name, gene number, coordinates of a gene, length of a corresponding protein, chain)
## HITS:3 COG:DRA0007 KEGG:DRA0007 NR:gi|6460585|gb|AAF12291.1|AE001862_117
(shows the number of homologs found in protein databases (takes into account maximum one
best homolog per a database), lists homologs IDs in the format DB:ID (e.g., COG:DRA0007);
notes:
- for homologs from NR, gi- numbers are given as homologs IDs;
- DB:ns indicates that a protein DB was not searched (e.g., NR:ns);
- DB:no indicates that a protein DB was searched but no homologs were found
(e.g., NR:no))
Then, complete ID lines of homologs are given preceded by DB names where they were found by BLAST (e.g., NR:) and followed by statistics from corresponding BLAST outputs.
## COG: DRA0007 COG1131 # Protein_GI_number: 15807679 # Func_class: V Defense mechanisms # Function: ABC-type multidrug transport system, ATPase component # Organism: Deinococcus radiodurans # 1 301 1 301 301 503 100.0 1e-142 ## KEGG: DRA0007 # Name: not_defined # Def: putative ABC-2 type transport system ATP-binding protein # Organism: D.radiodurans # Pathway: ABC transporters - General [PATH:dra02010] # 1 301 1 301 301 503 100.0 1e-142 ## NR: gi|6460585|gb|AAF12291.1| ABC transporter, ATP-binding protein, putative [Deinococcus radiodurans R1]^Agi|15807679|ref|NP_285331.1| ABC transporter, ATP-binding protein, putative [Deinococcus radiodurans R1] # 1 301 1 301 301 503 100.0 1e-141
BLAST parameters of similarity found for predicted protein are shown in the following order:
Start and stop of region of similarity ( 1 301) in predicted protein
Start and stop of region of similarity (1 301) in homolog from a database
Length of homologous protein (301)
BLAST score (503)
BLAST Identity (100.0 %)
BLAST Expected value (1e-141)
For genes predicted as a result of mapping of ribosomal proteins to sequences, description lines for predictions and ID lines of corresponding (predicted) proteins look like following (note "PROTEIN SUPPORTED" keyword):
164 86 Op 1 31/0.000 + CDS 189676 - 190599 1604 ## PROTEIN SUPPORTED gi|26246115|ref|NP_752154.1| 30S ribosomal protein S2 >gi|48994873|gb|U00096.2| GENE 164 189676 - 190599 1604 307 aa, chain + ## PROTEIN SUPPORTED ## NR: gi|26246115|ref|NP_752154.1| 30S ribosomal protein S2 [Escherichia coli CFT073] # 1 307 1 307 307 622 99 1e-176 LVSTTYLWYKARRTSDPFRIHRLDGSDNLTLCNNTHVSAHIPGCPLGSVIWDTWRHNPNF YIEVLIMATVSMRDMLKAGVHFGHQTRYWNPKMKPFIFGARNKVHIINLEKTVPMFNEAL AELNKIASRKGKILFVGTKRAASEAVKDAALSCDQFFVNHRWLGGMLTNWKTVRQSIKRL KDLETQSQDGTFDKLTKKEALMRTRELEKLENSLGGIKDMGGLPDALFVIDADHEHIAIK EANNLGIPVFAIVDTNSDPDGVDFVIPGNDDAIRAVTLYLGAVAATVREGRSQDLASQAE ESFVEAE
If you run mapping of ribosomal proteins to sequences with option "allow frameshifts / internal stop codons", predictions with frameshifts, if any, are noted with keyword "orf_shift", and predictions with internal stop codons, if any, are noted with keyword "orf_stop".
For example, here is a prediction with frameshift(s) (note "orf_shift"):
2 1 Op 2 11/0.000 + CDS 301 - 1973 2797 ## PROTEIN SUPPORTED orf_shift gi|24051180|gb|AAN42537.1| 30S ribosomal subunit protein S1 >904 GENE 2 301 - 1973 2797 557 aa, chain + ## PROTEIN SUPPORTED orf_shift ## NR: gi|24051180|gb|AAN42537.1| 30S ribosomal subunit protein S1 [Shigella flexneri 2a str. 301] # 1 557 1 557 557 1082 99 0.0 MTESFAQLFEESLKEIETRPGSIVRGVVVAIDKDVVLVDAGLKSESAIPAEQFKNAQGEL EIQVGDEVDVALDAVEDGFGETLLSREKAKRHEAWITLEKAYEDAETVTGVINGKVKGGF TVELNGIRAFLPGSLVDVRPVRDTLHLEGKELEFKVIKLDQKRNNVVVSRRAVIESENSA ERDQLLENLQEGMEVKGIVKNLTDYGAFVDLGGVDGLLHITDMAWKRVKHPSEIVNVGDE ITVKVLKFDRERTRVSLGLKQLGEDPWVAIAKRYPEGTKLTGRVTNLTDYGCFVEIEEGV EGLVHVSEMDWTNKNIHPSKVVNVGDVVEVMVLDIDEERRRISLGLKQCKANPWQQFAET HNKGDRVEGKIKSITDFGIFIGLDGGIDGLVHLSDISWNVAGEEAVREYKKGDEIAAVVL QVDAERERISLGVKQLAEDPFNNWVALNKKGAIVTGKVTAVDAKGATVELADGVEGYLRA SEASRDRVEDATLVLSVGDEVEAKFTGVDRKNRAISLSVRAKDEADEKDAIATVNKQEDA NFSNNAMAEAFKAAKGE
For other predictions (rRNA, promoters, etc.) we provide only description lines, for example:
- LSU_RRNA 884415 - 887254 98.0 # Leuconostoc oenos S60377
rRNAs are labeled as LSU_RRNA (large subunit), SSU_RRNA (small subunit) or 5S_RRNA (5S), tRNAs as TRNA, promoters as Prom, and terminators as Term.
Terminator regions (their coordinates and scores) are reported by BTERM program:
+ Term 492 - 537 -0.9
Promoters (their coordinates and scores) are reported by TSSB1 program.
c) How to run scripts of Fgenesb_annotator; configuration (paths) file
Main script - bamg.pl - runs all steps of training, gene prediction and annotation for a given sequence (or a set of sequences).
bamg.pl runs analysis for a set of sequences in FASTA format
Usage: bamg.pl
<paths file> - file with information about location of programs and options;
Three such files are included in the package:
ba_paths.list - to run on bacterial sequences
ar_paths.list - to run on archaebacterial sequences
ba_ar_paths.list - to run on unseparated mixture of bacterial + archaebacterial sequences
<output> - file where output will be saved;
<minlen> - minimal length of predicted genes in nucleotides;
<parameter file> - file with parameters for gene prediction (optional).
EXAMPLE: bamg.pl ba_paths.list test.set test.out 90
Description of lines in paths file
Example: ba_paths.list
# locations of programs and data for bacterial genome annotation /home/fgenesb/BACT_SCRIPTS/ ## directory with scripts / programs /home/fgenesb/BACT_SCRIPTS/bact.par ## 50 ## gene finding parameter file /home/blast-2.2.14/bin/blastall ## 8 ## 1 ## blast exe (for blastn) /home/fgenesb/DATA/rrna_db/bact_LSRRNA.set ## 1 ## 300 ## bacterial rRNA DB /home/fgenesb/DATA/rrna_db/bact_5SRRNA.set ## 1 ## 50 ## bacterial 5S rRNA DB /home/fgenesb/DATA/rrna_db/arch_LSRRNA.set ## 0 ## 50 ## archea rRNA DB /home/fgenesb/DATA/rrna_db/arch_5SRRNA.set ## 0 ## 50 ## archea 5S rRNA DB /home/fgenesb/tRNAscan/bin/tRNAscan-SE ## 2 ## 0(no) 1(b+a) 2(b) 3(a) /home/fgenesb/BACT_SCRIPTS/map.par ## parameters for protein mapping /home/blast-2.2.14/bin/blastall ## 10 ## 1 ## blast exe (for blastx) /home/fgenesb/DATA/rbp_db/nr_bact_rbp ## NR ## 1 ## 1 ## ribos. prot. DB /home/blast-2.2.14/bin/blastpgp ## 8 ## 1 ## blast executable /home/fgenesb/DATA/cog_db/cog.pro ## COG ## 1 ## COG database /home/fgenesb/DATA/cog_db/cog_gene.list ## COG gene list /home/fgenesb/DATA/cog_db/org.list ## COG organisms list 2 ## number of other prot. DB for BLAST /home/KEGG/kegg.fa ## KEGG ## 2 ## KEGG DB /home/NR/nr ## NR ## 2 ## NR DB 1 ## predict promoters/terminators 1 ## add sequence name to pred. gene
Some lines may be subdivided into fields (by separators ' ## ').
The same lines from "ba_paths.list" are shown in the table below in the 2nd column, with the numbers of lines indicated in the 1st column. Content of each line is explained after the table.
Note: in the example the number of other databases for BLAST is set to 2 and those databases are KEGG and NR. If you want to use more/other protein DBs for annotation, provide the number of DBs and list DBs with required parameters (name and BLAST mode) on the following lines.
1 |
# locations of programs and data for bacterial genome annotation |
2 |
/home/fgenesb/BACT_SCRIPTS/ ## dir. with programs/scripts |
3 |
/home/fgenesb/BACT_SCRIPTS/bact.par ## 50 ## gene finding parameters |
4 |
/home/blast-2.2.14/bin/blastall ## 8 ## 1 ## blast (for blastn) |
5 |
/home/fgenesb/DATA/rrna_db/bact_LSRRNA.set ## 1 ## 300 ## bact. rRNA DB |
6 |
/home/fgenesb/DATA/rrna_db/bact_5SRRNA.set ## 1 ## 50 ## bact. 5S rRNA DB |
7 |
/home/fgenesb/DATA/rrna_db/arch_LSRRNA.set ## 0 ## 50 ## arch. rRNA DB |
8 |
/home/fgenesb/DATA/rrna_db/arch_5SRRNA.set ## 0 ## 50 ## arch. 5S rRNA DB |
9 |
/home/fgenesb/tRNAscan/bin/tRNAscan-SE ## 2 ## 0(no) 1(b+a) 2(b) 3(a) |
10 |
/home/fgenesb/BACT_SCRIPTS/map.par ## parameters for protein mapping |
11 |
/home/blast-2.2.14/bin/blastall ## 10 ## 1 ## blast (for blastx) |
12 |
/home/fgenesb/DATA/rbp_db/nr_bact_rbp ## NR ## 1 ## 1 ## ribos. prot. |
13 |
/home/blast-2.2.14/bin/blastpgp ## 8 ## 1 ## blast executable |
14 |
/home/fgenesb/DATA/cog_db/cog.pro ## COG ## 1 ## COG database |
15 |
/home/fgenesb/DATA/cog_db/cog_gene.list ## COG genes list |
16 |
/home/fgenesb/DATA/cog_db/org.list ## COG organisms list |
17 |
2 ## number of other DBs for BLAST |
18 |
/home/KEGG/kegg.fa ## KEGG ## 2 ## KEGG DB |
19 |
/home/NR/nr ## NR ## 2 ## NR DB |
20 |
1 ## predict promoters/terminators |
21 |
1 ## add sequence name to pred. gene |
Content of each line:
1 - comments
2 - directory where FGENESB programs and scripts are located
3 - location of general gene finding parameter file;
second field - threshold number for predicted genes.
First, genes are initially predicted using general gene finding parameter file given in this line.
There are three such general parameter files in Fgenesb system (they were computed on mixtures of different microbial sequences):
bact.par - parameter file for bacterial sequences annotation
arch.par - parameter file for archaeal sequences annotation
gener.par - parameter file for mixed bacterial + archaeal sequences annotation
Second, if the number of predicted genes is more than the threshold number given in the line, then automatic training of gene finding parameters is involved and genes are re-predicted based on automatically generated parameters.
Otherwise, if the number of predicted genes is quite small (less than the threshold number), training parameters for gene prediction is not reliable and is not involved.
4 - location of BLAST executable (for BLASTN);
second field - E-value threshold (N means 1e-N, e.g., 8 means 1e-08);
third field - number of processors to use (value for BLAST '-a' option)
5 - location of bacterial 16S/23S rRNA database;
second field - use this database (1) or not (0);
third field - minimal length of a hit
6 - location of bacterial 5S rRNA database;
second field - use this database (1) or not (0);
third field - minimal length of a hit
7 - location of archaeal 16S/23S rRNA database;
second field - use this database (1) or not (0);
third field - minimal length of a hit
8 - location of archaeal 5S rRNA database;
second field - use this database (1) or not (0);
third field - minimal length of a hit
9 - location of tRNAscan-SE executable;
second field: 0 - do not use tRNAscan,
1 - use both bacterial and archaeal tRNA option,
2 - use only bacterial option,
3 - use only archaeal option
10 - location of file with thresholds and parameters for protein mapping
11 - location of BLAST executable (for BLASTX);
second field - E-value threshold (N means 1e-N, e.g., 10 means 1e-10);
third field - number of processors to use (value for BLAST '-a' option)
12 - location of (ribosomal) proteins DB to map proteins to genomic sequences;
second field - database name
third field - use the database (1) or not (0);
fourth field - allow frameshifts / internal stop codons in predicted proteins (1) or not (0)
13 - location of BLAST executable (blastpgp);
second field - E-value threshold (N means 1e-N, e.g., 8 means 1e-08);
third field - number of processors to use (value for BLAST '-a' option)
14 - location of COG database;
second field - database name (COG);
third field: 0 - do not BLAST predicted proteins against COG database,
1 - BLAST predicted proteins against COG database
15 - location of cog_gene.list file (list of COG genes)
16 - location of org.list file (list of organisms in COG)
17 - number of other protein databases for BLAST
Note: in this example the number of other databases for BLAST is 2 and those databases are KEGG and NR (lines 18, 19). If you want to use more or other protein DBs for annotation, put the number of DBs on this line and list DBs with required parameters (name and BLAST mode) on the following lines.
18 - location of KEGG database;
second field - database name (KEGG);
third field: 0 - do not BLAST predicted proteins against KEGG database,
1 - BLAST all predicted proteins against KEGG database,
2 - BLAST only those predicted proteins against KEGG database
that have no hits from other BLAST (against COG)
Note: when converting predictions from Fgenesb to Sequin format, gene names and EC numbers for predicted genes are assigned from KEGG homologs (if such are found) and protein names are assigned either from KEGG or NR homologs.
Therefore, if you are planning to submit genome annotation to GenBank, it is recommended to blast all predicted proteins against KEGG (mode 1 for KEGG).
19 - location of NR database;
second field - database name (NR);
third field: 0 - do not BLAST predicted proteins against NR database,
1 - BLAST all predicted proteins against NR database,
2 - BLAST only those predicted proteins against NR database
that have no hits from other BLASTs (against COG, KEGG)
20 - prediction of promoters / terminators: predict promoters / terminators (1) or not (0)
21 - adding names of sequences to ID lines of predicted proteins: add (1) or not (0)
Predictions with frameshifts / internal stop codons
When mapping proteins to sequences, ORFs with frameshifts or/and internal stop codons can be predicted. Predictions with frameshifts are noted with keyword "orf_shift", and predictions with internal stop codons are noted with "orf_stop".
If you intend to submit predictions to Genbank, there are several options to deal with predictions with frameshifts / internal stop codons:
1) run Fgenesb with option to NOT allow frameshifts / internal stop codons
for example, put in "ba_paths.list":
/home/fgenesb/DATA/rbp_db/nr_bact_rbp ## NR ## 1 ## 1 ## ribos. prot. DB
2) run Fgenesb with option to allow frameshifts / internal stop codons
for example, put in "ba_paths.list":
/home/fgenesb/DATA/rbp_db/nr_bact_rbp ## NR ## 1 ## 1 ## ribos. prot. DB
Then analyze ORFs with frameshifts / internal stop codons in Fgenesb output, adjust sequences of contigs so that there will be no such frameshifts / internal stop codons upon the next run, and re-run Fgenesb annotation on changed contigs.
Otherwise, if contigs are not adjusted and ORFs with frameshifts or/and internal stop codons are present in the output, converter "tosequin.pl" will skip such genes with errors upon conversion of predictions from Fgenesb to Sequin format.
d) Installation
Installation is simple, and the only problem that sometimes happens is that executables are not compatible with the user's operation system. The easiest solution is to open temporary account where we can compile, install and test FGENESB on your system.
Unpack the distribution files in a directory where you want to install the system. You will see the following directories:
BACT_SCRIPTS/ - FGENESB programs and scripts
DATA/ - COG, rRNA and bacterial ribosomal proteins databases
(subdirectories cog_db/, rrna_db/ and rbp_db/)
KEGG/ - scripts and description how to prepare KEGG database
TEST/ - example how to run FGENESB
TOGB/ - scripts to convert FGENESB predictions to GenBank format
TOSQ/ - scripts to convert FGENESB predictions to Sequin format
tRNAscan-SE-1.23/ - tRNAscan-SE program for detection of transfer RNA genes
(you can have tRNAscan-SE program from the authors or from our *.tar.gz file)
Install tRNAscan-SE
- go to tRNAscan-SE-1.23/ directory;
- find the following lines in Makefile:
BINDIR = $(HOME)/bin LIBDIR = $(HOME)/lib/tRNAscan-SE MANDIR = $(HOME)/man
and change $(HOME) to a directory where you want to install tRNAscan-SE, e.g.:
BINDIR = /home/fgenesb/tRNAscan/bin LIBDIR = /home/fgenesb/tRNAscan/lib MANDIR = /home/fgenesb/tRNAscan/man
- execute the following commands:
make make install
(see tRNAscan-SE-1.23/INSTALL for more details on how to install tRNAscan-SE)
BLAST and NR database
In addition to software/data from the distribution files, you will need to install BLAST software (and NR protein database if you are going to use it in annotation).
BLAST can be downloaded from NCBI web page:
http://www.ncbi.nlm.nih.gov/BLAST/download.shtml
Use BLAST release 2.2.13 or some higher version.
Download NR (non-redundant protein sequence database) from NCBI site:
a) you can download NR as a single FASTA file:
ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz
unpack 'nr.gz' and
format 'nr' for use with BLAST by 'formatdb' program (suppose, it is installed in /home/BLAST/):
/home/BLAST/formatdb -i nr -o T /home/BLAST/formatdb -i nr
(use either of 2 commands, '-o T' is optional)
b) alternatively, download NR from NCBI as BLAST preformatted files:
ftp://ftp.ncbi.nih.gov/blast/db/
nr.00.tar.gz
nr.01.tar.gz
nr.02.tar.gz
KEGG database
See "KEGG_prepare.txt" from FGENESB/KEGG/ directory for how to prepare and install KEGG database for use with FGENESB.
COG, rRNA and ribosomal proteins databases
In case if included COG, rRNA and ribosomal proteins blast-formatted files do not work properly with BLAST on your computer, execute formatdb with COG, rRNA, ribosomal proteins databases:
/home/BLAST/formatdb -i <datafile> -p F
where <datafile> is each of the following files:
arch_5SRRNA.set arch_LSRRNA.set bact_5SRRNA.set bact_LSRRNA.set /home/BLAST/formatdb -i cog.pro /home/BLAST/formatdb -i nr_bact_rbp
Configuration file (paths file)
Three examples of configuration files are given in BACT_SCRIPTS/ and TEST/ directories:
ba_paths.list - for annotation of bacterial genomes ar_paths.list - for annotation of archaebacterial genomes ba_ar_paths.list - for annotation of mixed bacterial + archaebacterial seq.
Edit configuration files - provide paths to software/data you have installed and options to run FGENESB system.
Running
go to TEST/ directory and run:
../BACT_SCRIPTS/bamg.pl ba_paths.list test.seq test.out 90
Here:
bamg.pl - main FGENESB script ba_paths.list - configuration file test.seq - test sequence test.out - output file 90 - minimal length of predicted genes in nucleotides.
The system is installed correctly if the output file 'test.out' is similar to 'test.res' (located in TEST/ directory) and no ERROR messages appear.
Now you can run the system on your sequence or a set of sequences, e.g., by replacing 'test.seq' with an appropriate file in the command above.
Note:
if you want to run several annotations in parallel, run each sequence, or a set of sequences, from a separate directory.
Gene finding parameters
If you make predictions in short contigs, gene finding parameters training procedure is not involved (because there is not enough data for training). In this case, either generic or pretrained gene finding parameters can be used for gene prediction.
If training procedure is not involved, generic gene finding parameters are used by default. Three generic gene finding parameters files suitable for annotating bacterial communities are included in FGENESB system:
bact.par (for bacteria) arhae.par (for archaea) gener.par (for bacteria + archaea)
You should indicate one of them in configuration file (for example, in "ba_paths.list").
On the other hand, you might want to use specific pretrained gene finding parameters obtained from related bacteria or large contigs of a query genome. In this case, provide pretrained gene finding parameters file with '-p' option to "bamg.pl" script:
bamg.pl <paths file> <sequence set> <output> <minlen> <-p param_file> <-cN>
where
-p - command to use pretrained parameters (optional) <param_file> - pretrained gene finding parameters file -c - command to use alternative genetic code N - number of genetic code (default is 11)
EXAMPLE:
../BACT_SCRIPTS/bamg.pl ba_paths.list test.seq test.out 90 -p dein.par -c4
You can compute your own parameters file using mgpa.pl script:
mgpa.pl <sequence set> <output> <param_file> <directory>
where
<sequence set> - fasta formatted sequence(s) <output> - output file with predictions <param_file> - file with trained parameters <directory> - directory with mgpa.pl script
EXAMPLE:
../BACT_SCRIPTS/mgpa.pl test.seq test.out test.par ../BACT_SCRIPTS/
Preliminary predictions
If you have pretrained parameters, you can run 'morfso' and get preliminary predictions (no tRNA, rRNA genes predictions, no ribosomal proteins mapping, no search for homology to COG, KEGG, or NR proteins, etc.) just in a minute, e.g.:
../BACT_SCRIPTS/morfso test.par test.seq 90 > NC_001264.res
(90 is a minimal length of ORF and is a required parameter)
e) Conversion to GenBank format
FGENESB annotation can be converted to GenBank format by scripts from TOGB/ directory.
Usage: run_tgb.pl <fgenesb_annotation_file> header <sequence_set> > <genbank_file>
<sequence_set> - file with fasta sequences; <fgenesb_annotation_file> - their annotation by Fgenesb
"header" is file with GenBank Header - you can edit it to put your names and other specific information, or you can use this file without editing but later edit *.gb file.
Example: run_tgb.pl test.res header test.seq > test.gb
See example of FGENESB output in GenBank format in "Explanation of Fgenesb_annotator output".
f) Conversion to Sequin format
FGENESB annotation can be converted to Sequin format (for submission to GenBank, after annotation is complete) by scripts from TOSQ/ directory. See "tosequin.txt" from TOSQ/ for details.
g) Appendix. Running gene/operon prediction steps in manual mode. Actual scripts.
As an alternative to automatic annotation, you can run each step separately in manual mode. (Here exemplified by annotation of NC_001264.fna).
STEP 1.
Finds all potential ribosomal RNA genes using BLAST against precompiled bacterial and/or archaeal rRNA databases. First, given genomic sequence is blasted against defined rRNA databases
and rRNA genes are detected. Then detected rRNA genes are masked so that they do not interfere with subsequent prediction of protein-coding genes. At a later stage (see bactg_ann.pl) scripts rna_annot.pl (for bacterial rRNA) and ra.pl (for archaeal rRNA) put non-redundant, non-overlapping rRNA genes in the final annotation file.
STEP 2.
Predicts tRNA genes using tRNAscan-SE program. Detected tRNA genes are masked so that they do not interfere with subsequent prediction of protein-coding genes. At a later stage (see bactg_ann.pl) script trna_annot.pl puts predicted tRNA genes in the final annotation file.
STEP (after 2, before 3). Mapping bacterial ribosomal proteins to genomic sequences and selecting good mappings.
STEP 3.
mgpa.pl makes parameter file and makes first prediction, without predicting operons (iterative procedure).
Usage: mgpa.pl <sequence> <output> <param.output> <directory> [codon (optional)]
Requires: orfs0, morfs, sc3_au, cod6m, codpotm, le_au, lexs0, prorf.pl, cmp2.pl
orfs0 - initial prediction of long slightly overlapping ORFs in sequence which are used as a starting point for calculating parameters of predictions;
morfs - ab initio gene prediction using precomputed parameters such as 5th-order in-frame Markov model for coding regions, averaged 2nd-order Markov model for region around start codon and upstream RBS site and probability distributions of ORF lengths.
mgpa.pl uses only genome sequence and optionally non-canonical genetic code specified as the translation table number at NCBI, default is 11 (canonical code). Of 86 annotated bacterial genomes, 81 have standard code with three stop codons (code 11), and five, for example M.genitalium and M.pneumonia, have two stop codons (code 4).
Script has two output files: <output> which is similar to gene prediction output of FGENESH (gene coordinates and predicted proteins at the end) and <param.output> which is output file for parameters that can be used for future gene predictions on this and related genomes.
3 EXAMPLE: mgpa.pl NC_001264.fna NC_001264.stp3 NC_001264.par /home/fgenesb/STEPS/
STEP 4.
morfso - using parameter file, makes predictions (including prediction of operons/TU)
Usage: morfso <param> <sequence> <min_length> [code: "-c X", optional, default = 11] > output
<min_length> - minimal length of predicted genes in nucleotides
This is a first run of morfso in which it predicts operons based only on distances between genes.
4 EXAMPLE: morfso NC_001264.par NC_001264.fna 100 > NC_001264.stp4
STEP 5.
Runs BLAST for predicted proteins against COG database (cog.pro).
Usage: runblast.pl <prot file> <DB> <output file> <directory> <blast> <blast(1), sbl(0)> <E-value> <proc number>
where:
<prot file> - file with proteins for BLAST <DB> - database, in this case - COG <output file> - output file with BLAST (parsed) results <directory> - directory with FGENESB programs/scripts <blast> - location of BLAST / DBscan executable <blast(1), sbl(0)> - 1 - use BLAST, 0 - use DBscan <E-value> - E-value threshold (N means 1e-N, e.g., 10 means 1e-10) <proc number> - number of processors to use for BLAST (value for BLAST -a option)
Requires: blastparse.pl or sblparse.pl
5 EXAMPLE: take_prot.pl NC_001264.stp4 NC_001264.stp4.prot (take proteins from predictions)
runblast.pl NC_001264.stp4.prot /home/fgenesb/DATA/cog_db/cog.pro NC_001264.stp5 /home/fgenesb/STEPS/ /home/BLAST/blastpgp 1 10 1
Step 5 is optional, if you do Step 5, then do Steps 6,7, otherwise skip them.
STEP 6.
oppr.pl - finding conserved operonic pairs from blast output through cog data.
Usage: oppr.pl <output from runblast.pl> <genes list> <organisms list> > output
Makes output with pairs of adjacent genes in the same strand which also occured adjacently in other 43 cog genomes, and puts number of occurences of pairs in the genomes and P-value, probability of observing by random chance. It uses two files: genes list - cog_gene.list and organisms list - org.list.
6 EXAMPLE:
oppr.pl NC_001264.stp5 /home/fgenesb/DATA/cog_db/cog_gene.list /home/fgenesb/DATA/cog_db/org.list > NC_001264.stp6
STEP 7.
Usage: morfso <param> <sequence> <min_length> -o <output from oppr.pl> > output
Second run of morfso which now uses information about conserved pairs to improve operon prediction.
7 EXAMPLE: morfso NC_001264.par NC_001264.fna 100 -o NC_001264.stp6 > NC_001264.stp7
STEP 8.
Runs BLAST for predicted proteins against KEGG database.
Usage: runblast.pl <prot file> <DB> <output file> <directory> <blast> <blast(1), sbl(0)> <E-value> <proc number>
where:
<prot file> - file with proteins for BLAST <DB> - database, in this case - KEGG <output file> - output file with BLAST (parsed) results <directory> - directory with FGENESB programs/scripts <blast> - location of BLAST / DBscan executable <blast(1), sbl(0)> - 1 - use BLAST, 0 - use DBscan <E-value> - E-value threshold (N means 1e-N, e.g., 10 means 1e-10) <proc number> - number of processors to use for BLAST (value for BLAST -a option)
Requires: blastparse.pl or sblparse.pl
8 EXAMPLE: take_prot.pl NC_001264.stp4 NC_001264.stp4.prot (take proteins from predictions)
runblast.pl NC_001264.stp4.prot /home/KEGG/kegg.fa NC_001264.stp8 /home/fgenesb/STEPS/ /home/BLAST/blastpgp 1 10 1
STEP 9.
Runs BLAST for predicted proteins against NR database.
Usage: runblast.pl <prot file> <DB> <output file> <directory> <blast> <blast(1), sbl(0)> <E-value> <proc number>
where:
<prot file> - file with proteins for BLAST <DB> - database, in this case - NR <output file> - output file with BLAST (parsed) results <directory> - directory with FGENESB programs/scripts <blast> - location of BLAST / DBscan executable <blast(1), sbl(0)> - 1 - use BLAST, 0 - use DBscan <E-value> - E-value threshold (N means 1e-N, e.g., 10 means 1e-10) <proc number> - number of processors to use for BLAST (value for BLAST -a option)
Requires: blastparse.pl or sblparse.pl
9 EXAMPLE: take_prot.pl NC_001264.stp4 NC_001264.stp4.prot (take proteins from predictions)
runblast.pl NC_001264.stp4.prot /home/NR/nr NC_001264.stp9 /home/fgenesb/STEPS/ /home/BLAST/blastpgp 1 10 1
STEP 10.
Adds names of homologs from COG/KEGG/NR (found through BLAST) to annotation file
(file with prediction results).
Usage: add_hml_info.pl <gene pred output> <nr_list> (<BLAST_MODE(0|1|2)> <blast output>)x3 (for COG/KEGG/NR) > output
where:
<gene pred output> - gene prediction output file
<nr_list> - location of "nr.list" file (names of proteins from NR database)
<BLAST MODE> - 0, 1, or 2:
0 - there was no BLAST of predicted proteins against protein database;
1 - there was BLAST of all predicted proteins against protein database;
2 - there was BLAST of only those predicted proteins against protein database
that had no hits from previous BLASTs
<blast output> - output file with BLAST (parsed) results
Last two parameters, <BLAST MODE> and <blast output>, must be repeated 3 times to indicate parameters for COG, KEGG, NR, respectively.
10 EXAMPLE:
add_hml_info.pl NC_001264.stp7 /home/NR/nr.list 1 NC_001264.stp5 1 NC_001264.stp8 1 NC_001264.stp9 > NC_001264.stp10
where NC_001264.stp5 - (parsed) BLAST output for COG NC_001264.stp8 - (parsed) BLAST output for KEGG NC_001264.stp9 - (parsed) BLAST output for NR
If BLAST was not used for any database (steps 5-9 were skipped), instead of using "add_hml_info.pl", just copy:
cp NC_001264.stp4 NC_001264.stp10
STEP 11.
pta.pl - predicts potential promoters (tssb1) or terminators (bterm) in corresponding 5'-upstream
and 3'-downstream regions of predicted genes, if there are no close genes in the neighborhood at the same strand. After that puts predicted promoter and terminator positions in the annotation file.
tssb1 - bacterial promoter prediction (sigma70) using discriminant function with characteristics of sequence features of promoters (such as conserved motifs, binding sites, etc.).
bterm - prediction of rho-independent terminators as hairpins, with energy scoring based on discriminant function of elements of hairpin.
Usage: pta.pl <directory> <genome seq> <gene pred file> > output
11 EXAMPLE: pta.pl /home/fgenesb/STEPS/ NC_001264.fna NC_001264.stp10 > NC_001264.stp11
STEP 12.
ref_opp.pl - refinement of operon predictions in the annotated file, using predicted promoters and terminators as an additional evidence.
Usage: ref_opp.pl <bacterial annotated file> > output
12 EXAMPLE: ref_opp.pl NC_001264.stp11 > NC_001264.res
Example: running steps separately in manual mode
(exemplified by annotation of seq. NC_001264.fna).
Copy programs/scripts to STEPS/ directory if you want to run annotation is steps, e.g., /home/fgenesb/STEPS/. Many steps are optional. Here we give one variant of running the pipeline in steps.
[Commands for first two steps, prediction of rRNA and tRNA genes, are not shown here.]
3 EXAMPLE: mgpa.pl NC_001264.fna NC_001264.stp3 NC_001264.par /home/fgenesb/STEPS/ 4 EXAMPLE: morfso NC_001264.par NC_001264.fna 100 > NC_001264.stp4 5 EXAMPLE: take_prot.pl NC_001264.stp4 NC_001264.stp4.prot
runblast.pl NC_001264.stp4.prot /home/fgenesb/DATA/cog_db/cog.pro NC_001264.stp5 /home/fgenesb/STEPS/ /home/BLAST/blastpgp 1 10 1
6 EXAMPLE:
oppr.pl NC_001264.stp5 /home/fgenesb/DATA/cog_db/cog_gene.list /home/fgenesb/DATA/cog_db/org.list > NC_001264.stp6
7 EXAMPLE: morfso NC_001264.par NC_001264.fna 100 -o NC_001264.stp6 > NC_001264.stp7 8 EXAMPLE: take_prot.pl NC_001264.stp4 NC_001264.stp4.prot
runblast.pl NC_001264.stp4.prot /home/KEGG/kegg.fa NC_001264.stp8 /home/fgenesb/STEPS/ /home/BLAST/blastpgp 1 10 1
9 EXAMPLE: take_prot.pl NC_001264.stp4 NC_001264.stp4.prot
runblast.pl NC_001264.stp4.prot /home/NR/nr NC_001264.stp9 /home/fgenesb/STEPS/ /home/BLAST/blastpgp 1 10 1
10 EXAMPLE:
add_hml_info.pl NC_001264.stp7 /home/NR/nr.list 1 NC_001264.stp5 1 NC_001264.stp8 1 NC_001264.stp9 > NC_001264.stp10
11 EXAMPLE: pta.pl /home/fgenesb/STEPS/ NC_001264.fna NC_001264.stp10 > NC_001264.stp11 12 EXAMPLE: ref_opp.pl NC_001264.stp11 > NC_001264.res
These steps require only a sequence file as an input, so it is easy to put them in a batch file and run by a single command, as we do using main script bamg.pl, but doing annotation in steps makes spotting errors easier and also allows to use non-standard genetic code.