Services Test Online
Note. To run applet viewer you need to include www.softberry.com in the list of your browser (see instruction).
Softberry GenomeSequence Explorer
Short Help
Softberry Human GenomeSequence Explorer help (pdf)
Softberry GenomeSequence Explorer allows visualization of many features, such as known and predicted genes, mRNAs, EST, promoters and many others, on large genomic sequences. On this server, GenomeSequence Explorer shows these features for human genome draft from UCSC Human Genome Project Team. In addition to publicly available information, GenomeSequence Explorer includes genes predicted by Softberry's FGENESH++ program. GenomeSequence Explorer can be used to extract nucleotide and protein sequences of particular genes, functional sites and other features. These sequences can be viewed by right-clicking features of interest in Gene Explorer's main window.
GenomeSequence Explorer is not as fast through the Internet as it is locally, so please be patient.
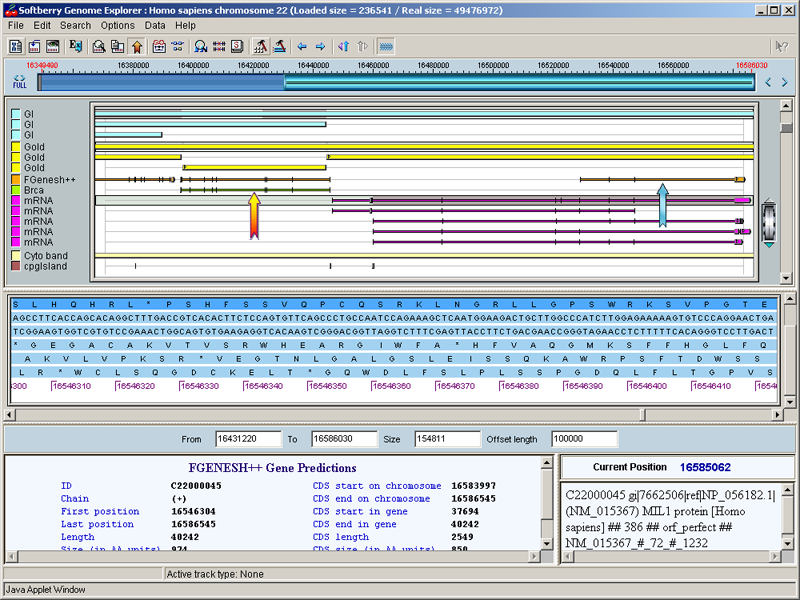 |
Click here for use restrictions.
Searching Genome for Feature of Interest
GenomeSequence Explorer has search function that helps you visualize features of interest on a genome. Features can be searched by GenBank AC number (mRNAs and known genes), EST ID, cytogenetic band name, repeat name or FGENESH++ predicted gene ID. Search can be performed only on feature types loaded into memory (Options > Load Feature Types option, see below). To start search, press FIND FEATURE button or select Search > Find Feature option from a menu. Find Feature dialog is displayed, where you can specify search conditions. After search was performed, a list of found features is displayed. To view a feature and its surroundings on genome map, click on a feature from the list and press LOAD button. List of found features is stored in memory until new search is performed or GenomeSequence Explorer is exited.
Selecting Chromosome Region.
Chromosome can be selected from Chromosome Selection Dialog Box. To open it, either choose Options> Chromosome from main menu or press Chromosome button. In Chromosome Selection Dialog Box, select chromosome from pull-down list. Then select chromosome region to be loaded. It can be done using a ruler just below the pull-down list, or by entering selected sequence boundaries' positions in fields FROM, TO and/or SIZE. By default, a 600-kb fragment of chromosome 1 is loaded. Please keep in mind that the larger selected region, the longer it will take to load and the slower to navigate.
The ruler is a graphical representation of entire given chromosome. You can choose region to load by moving and reshaping selection (green shaded rectangle) along its length:
- Dragging selected region by mouse to the left or to the right;
- Dragging one of the region's boundaries;
- Shift-dragging region's boundary - in this case both boundaries expand or retract to the same extent.
Boundaries of selected region are displayed in fields FROM and TO below chromosome's map, and its size in SIZE field. To select by sequence position, enter start and end positions in the fields FROM and TO, or start (end) position and length in fields FROM (TO) and SIZE. Press ENTER key or move cursor to another field to record entered values. If entered values in any of the fields go beyond chromosome's boundaries, they are corrected and new values are displayed in the fields. Changing value in one of the field may result in change in another - e.g. entering new value in FROM will change value in SIZE. Selected region may be shifted to the left or to the right by pressing buttons <
Selecting Features to Display.
Options > Load Feature Types menu option allows you select features to be loaded in computer memory. Of these loaded, some or all can be displayed in GenomeSequence Explorer window. Selecting Features to display can be done by either pressing FEATURES button or choosing Options> Features from main menu. The following features can be selected:
- Gap locations (on nucleotide sequence)
- Golden Path Positions of chromosome segments
- Gold chromosome assembly
- Genes predicted by FGENESH++ program
- Known genes (from GenBank/Infogen entries)
- Known mRNA sequences
- Known EST sequences
- SNPs from clone overlaps
- SNPs from random reads
- Sequence Tag Marker locations
- Repeat locations
- Mouse syntheny regions
- Chromosome cytogenetic band locations.
By default all features are selected in both LOAD FEATURES TYPES and FEATURES dialogs. Search can be performed only among loaded features.
Navigating Main Window
After choosing chromosome region and features, a fragment of loaded region is displayed in the main window. You can change size and location of displayed fragment using ruler and boundary/size fields the same way you did it in Chromosome Selection Dialog Box. But in the main window, a ruler represents loaded chromosome region rather than whole chromosome.
You can also move along the region's length by dragging main window map by mouse to the left or to the right. Shift-drag to the left decreases map resolution, while same move to the right increases it. If option "Repaint on Drag" is enabled (default), the map moves along with mouse. If it is disabled, map is redrawn only after mouse button is released. Finally, you can choose a region on the map by Ctrl-dragging, and this region will expand over whole window. Maximal resolution of GenomeSequence Explorer is one base pair per pixel.
Features of selected region are displayed in the middle section of main window. Each feature type is shown on its own line(s). By default, if features of same type overlap on sequence, they are shown on separate lines, while top line for each type of features (shaded in gray) shows all features of that type combined. You can switch off display of combined lines or separate lines by choosing Options > Feature Show Mode > All Objects or > Combined Objects.
When mouse pointer touches some feature on the map, it changes its shape to a hand. At that time, if option "Show Information on Mouse Over" is enabled (default), available information on each feature is shown in left bottom panel (Feature Info Panel). If that option is disabled, you must click left button to get information displayed. If mouse pointer is located over multi-block (multi-exon) feature, like gene or EST with several alignment blocks, Feature Info Panel is divided into two sections. In left section, information about whole section is displayed, while in right section, information about selected (pointed to) block.
On top of main window's ruler, there are buttons "Back" and "Forward" which operate in a way similar to same buttons in Internet browsers.
Mouse right-clicking anywhere on the map opens a menu with the following options:
- Show Sequence - (+) and (-) - Shows nucleotide sequence around the point clicked, positive or negative strand.
- Show Feature Sequence - (+) and (-) - Shows nucleotide sequence of a given feature on either of two strands, as well as short flanks. Enabled only when pointer is located over a feature (hand-shaped).
- Show Block Sequence - (+) or (-) - shows nucleotide sequence of a given block (exon).
- Show Protein - Shows amino acid sequence of gene product. Enabled only if gene is pointer is located over a gene.
- Show Entry - Shows GenBank entry for mRNA or EST.
Sequences and entries are shown in a separate window. Options of sequence display are determined by Sequence Options menu (Options>Sequence). Sequences of selected features are shown in bold capital letters, while flanks are shown in lowercase. Exons on exon-containing features (gene, mRNA, EST), are highlighted in blue. Selected exons, if any, are shown in red.
Menu Descriptions.
File menu:
- Exit - exit the viewer.
Search menu:
- Find Feature - opens dialog for feature search.
- Found Feature List - shows last feature search results.
Options menu:
- Chromosome - Choose chromosome to view.
- Features - Choose features to view (only those loaded by Load Feature Type can be viewed).
- Load Feature Types - choose features to load in computer memory.
- Sequence - Select sequence display options: flank lengths for feature sequence display, length of sequence to be displayed, numbering options etc.
- Repaint on Drag - If enabled (default), view is updated continuously when drag operation is performed by mouse. If disabled, view is updated only when drag operation is finished (mouse button released).
- Show Information on Mouse Over - If enabled (default), information on functional site is displayed if mouse is located over that site. If disabled, information is displayed after a click on a functional site.
- Show Navigation Line (default enabled) - displays vertical line at the location of mouse pointer.
- Allow Visual Map Navigation (default enabled) - allows navigation by dragging map by mouse to the left or to the right.
- Feature Show Mode - choose between displaying overlapping features of same type on separate lines (All Objects), on one line (Combined Objects) or both. Also, Full Mode displays each individual known overlapping EST or mRNA on a separate line, while Compact Mode displays only one EST/mRNA of a class.
Use Restrictions.
Data in this viewer were generated by Softberry or copied from publicly available genome database information of UCSC Human Genome Browser. All users are wellcome to review this information, but some parts of it cannot be used for commercial purposes without license from Softberry or UCSC. Full set of human genome genes as predicted by FGENESH++ is distributed exclusively by Biomax Informatics AG (www.biomax.de) and should be licensed from them