Services Test Online
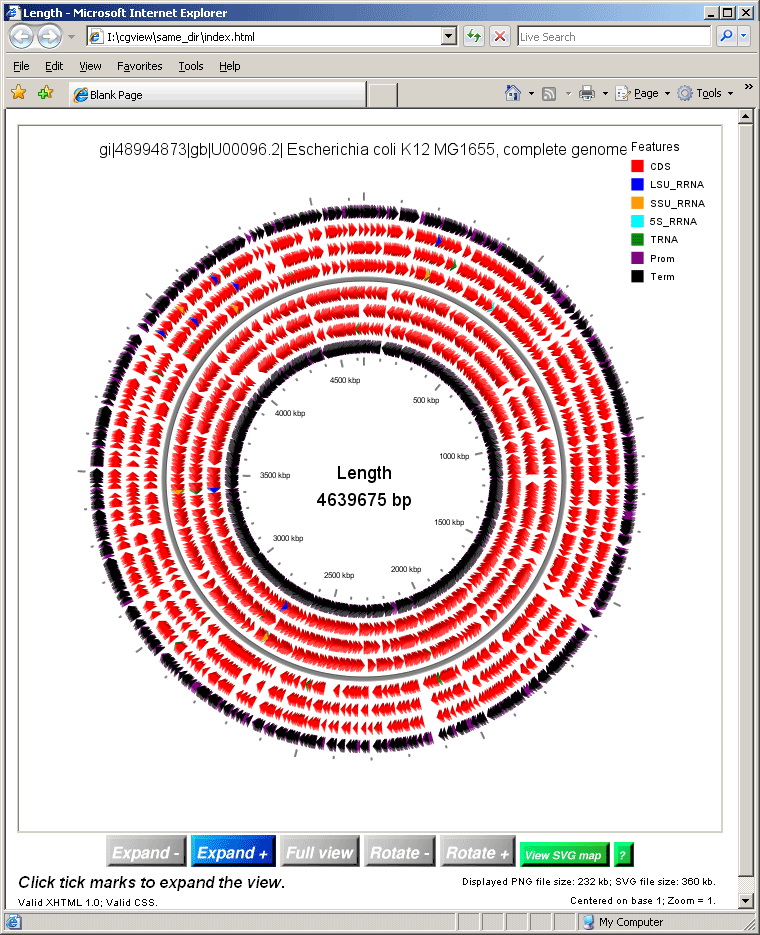
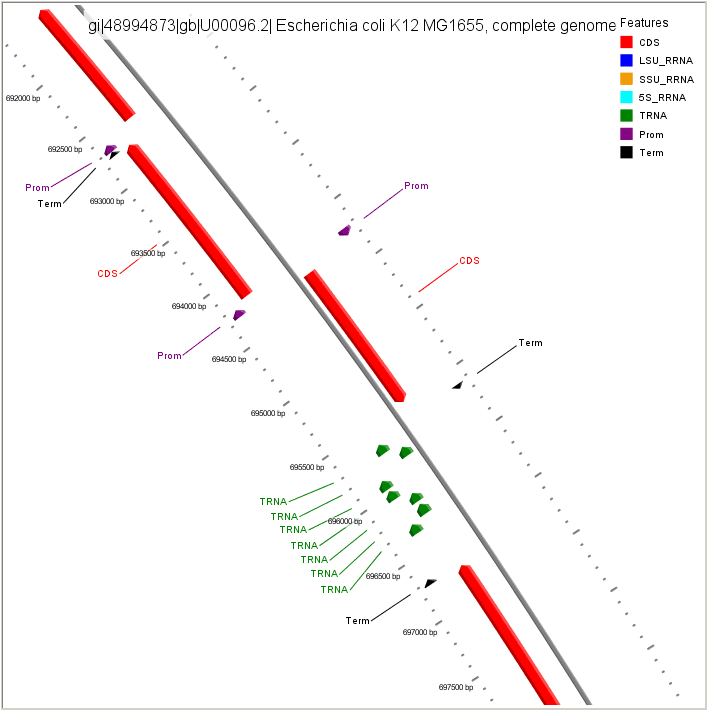
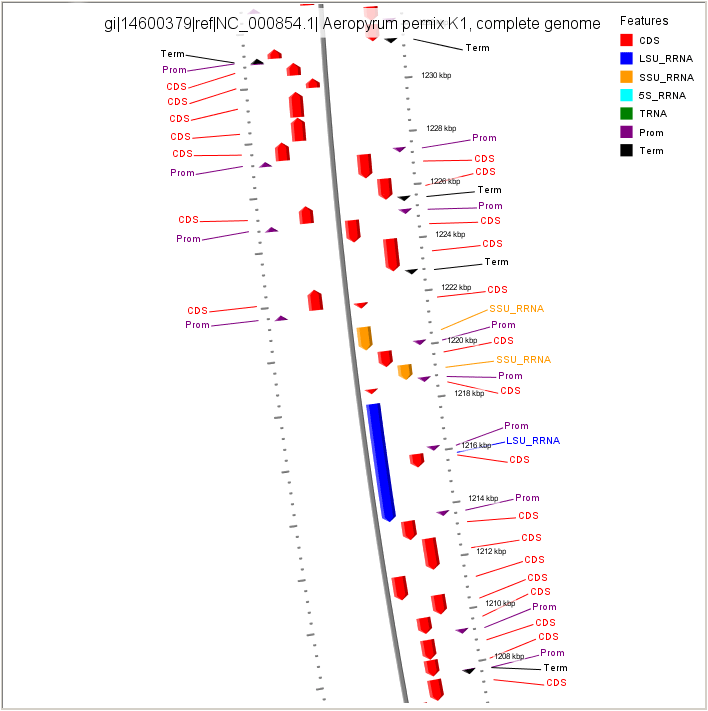
Visualization of FgenesB annotation using CGView
CGView software developed by: Stothard P, Wishart DS. Circular genome visualization and exploration using CGView. Bioinformatics 21:537-539.
TOCGVIEW perl script serves to convert data from the FgenesB annotator pipeline format to CGView xml format.
To run TOCGVIEW click here (WEB version of visualization has less options than running locally and uses only standard set of features names)
You can download TOCGVIEW here .
Options:
| --input | Input file in FgenesB format |
| --output | Output file |
| --feature |
The list of visualized features (separated by comma if more than 1). By default - all
CDS - protein coding gene LSU_RRNA - rRNA genes for large subunit SSU_RRNA - rRNA genes for small subunit 5S_RRNA - rRNA genes for 5S subunit TRNA - tRNA genes Prom - Promoters Term - Terminators |
| --exclude | List of features not to visualize (separated by comma if more than 1) |
| --noframes | Do not split genes into frames, show them just as forward and reverse genes |
| --format |
Output format type:
xml (default) - http://wishart.biology.ualberta.ca/cgview/xml_overview.html tab - http://wishart.biology.ualberta.ca/cgview/tab_input.html |
| --help | Print help text and exit |
EXAMPLES of local running above mentioned scripts for visualization:
perl tocgview.pl --input Fgenesb.ann --output out.xml
In this example all features will be processed.
perl tocgview.pl --input Fgenesb.ann --output out.xml --feature Term,Prom
In this example will be processed terminators and promoters only.
Input file example
(file in FgenesB format)
Output file example
In order to start tocgview and CGView
1) run tocgview.pl to generate cgview xml file.
For example: perl tocgview.pl --input Fgenesb.ann --output out.xml
2) run cgview.jar for visualization. Read
http://wishart.biology.ualberta.ca/cgview/application.html for getting help.
It makes sense to run cgview with "-s" option.
For example: java -jar cgview.jar -i out.xml -s same_dir