Services Test Online
ReadsClean: The Package for Purging Random Errors from Large Volumes of Short Sequences (reads)
CONTENTS
Description
Synopsis
Build
Commands And Options
Sample
License And Citation
Downloads
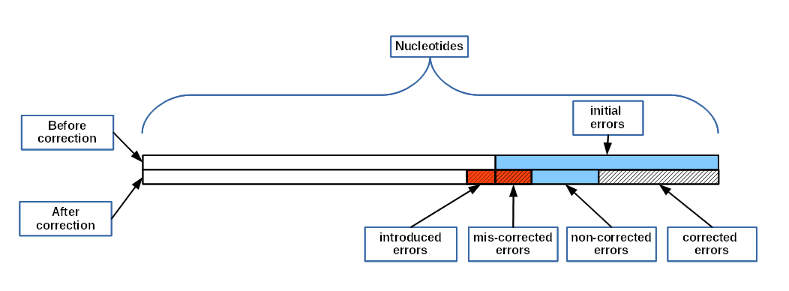
"left errors" = "non-corrected errors" + "wrong-corrected errors"
"wrong-corrected errors" = "introduced errors" + "mis-corrected errors"
"changed" = ("initial errors" - "non-corrected errors") + "introduced errors"
Where:
Nucleotides - total number of nucleotides in simulated reads
leftover errors - total number of errors left after cleaning
non-corrected errors - missed errors
wrong-corrected - wrongly corrected nucleotides (mis-corrected plus introduced errors)
introduced errors (over-corr) - errors introduced during cleaning
mis-corrected - initial incorrect nucleotide replaced by another incorrect nucleotide
changed - total number of changes during cleaning (corrected errors plus wrongly corrected)
initial errors - number of errors in reads before cleaning
corrected errors - true corrected errors
Description
readsClean is a software package for purging random errors from large volumes of short sequences (reads), while keeping nonrandom mutations (SNPs).
Synopsis
Clean end bases of paired reads, use eight processors and set a work folder as ./cleandir
Step 1. Create configuration file clean.cfg with the following content:
reads.pe.dt {
--cleaning
src.file = "reads.fa"
src.format = "ifasta-pair"
aver_pe=200
stdev_pe=30
}
hardware {
memory.max = 8
memory.opt = 4
}
Step 2. Run a cleaner script:
./bin/clean.pl -cfg:./bin/cfg/clean.def.cfg -cfg:./clean.cfg -work_dir:./cleandir -j:8
An output will be saved in a ./cleandir/res folder
Build
Run make_all.pl --release in a ./src folder.
Commands And Options
Specify configuration files
-cfg:fname Main settings are specified in configuration files. Default settings are stored in cfg/assembling.def.cfg, this file must be entered in a command line before users’ own configuration file. User can also create and use his own configuration file(s) (user.cfg in an example), which would contain paths to input files and user-defined (see section CONFIGURATION FILES). User-specified configuration file must always follow default file in a command line.
Work folder.
-work_dir:XX Set a work folder. An output will be saved in a subfolder XX/res.
Multiprocessing.
-j:XX Sets a number of processes for multiprocessing regime.
CONFIGURATION FILES
The input data sources are specified in a configuration file, which can contain one or several sections, each set of reads being described in its own section. Below is an example of a section specifying input reads, with explanation of keys:
{
--cleaning:on
--clean-by-self:off
--clean-other:off
--clean-by-other:off
--stat-base
src.file = "reads.fa"
src.format = "ifasta-pair" {
def_fasta_qua = 25
phread=64
}
aver_pe=200
stdev_pe=30
}
Name of each section, which specifies characteristics of an individual read set, consists of three identifiers separated by a period (.) First identifier for this type of section is always "reads".
Next identifier defines type of pairwise reads:
.se - "SE" reads type – single reads .pe - "PE" reads type – "PE" pair-end reads .mp - "MP" reads type – "MP" pair-end reads
Third identifier is a unique name of a given set. Any combination of letters and numbers can be used. So, a section with name reads.pe.dt defines parameters for "PE" pair-end reads.
Each section specifies keys that determine the parameters of operation with reads. The keys assume values “on” and “off”. If a value is missing it is assumed to be “on”. Therefore, record –cleaning:on is equivalent to а –cleaning.
--cleaning:on - use the read set in cleaning. If turned off (--cleaning:off), the reads from that set won’t be used in cleaning.
--clean-by-self:on - cleans read set using reads from the same set. To turn on: --clean-by-self:off.
--clean-by-other:on - Clean given read set using reads from other sets.
--clean-other:on - Clean other read sets using reads from this set.
--stat-base - Outputs some statistics for given read set in a file in subdirectory /res of work directory.
src.format = "fasta-pair" – Format of a file containing reads. Allowed formats:
- fasta-pair – FASTA file where a second read for each pair (even) immediately follows a first (odd).
- fasta – for sections of se type, FASTA file with SE reads. For sections of pe or mp types, two files shall be specified, where first reads for each pair are recorded in a first file, and second reads – in a second file in the same order.
- fastq-pair – FASTQ file with pairwise reads directly following each other (even, odd).
- fastq – for sections of se type, FASTQ file with SE reads. For sections of pe or mp types, two files shall be specified, where first reads for each pair are recorded in a first file, and second reads – in a second file in the same order.
src.file = "reads.fa" – efines path to file(s) with reads (either absolute or relative paths). For pairwise reads separated in two files, two paths shall be entered inside square parentheses: src.file = ["reads1.fa" "reads2.fa"].
aver_pe=200 - Average distance between reads.
stdev_pe=30 - Standard deviation of that distance.
The program outputs reads sorted into two categories:
1. Reliably cleaned ("clean subset") – percentage of errors is reliably low .
2.
Reads not reliably cleaned ("dirty subset") – reads that cannot be verified and/or cleaned along their entire lengths (due to low coverage multiple and/or high rate of errors). Accordingly, the percentage of errors in this subset is very high.
--single-out:on - Save all results in a single file, i.e. not separate them into files containing cleaned and "dirty" subsets.
- If this key is turned off (default setting) only reliably cleaned reads are saved - and sorted into two files:
- identifier .pe.pair – Cleaned reads that retained a pair (both reads of a pair were reliably cleaned).
- identifier .se.single – Cleaned reads that lost their pair (only one read was cleaned reliably).
If this key is turned on (--single-out:on), all reads - reliably cleaned and "dirty" - are saved in the same file.
Hardware section lets user set the size of memory used by the software – in whole Gb.
hardware {
memory.opt = 4
memory.max = 8
}
The parameter memory.opt sets the size of memory allocated to the software, while memory.max – sets an absolute upper limit of memory that can be used. Obviously, the larger size of used memory, the faster the software processes the data.
Example
This is an example of configuration file where reads from reads1.fa file are cleaned by reads from reads2.fa, while the latter file itself is not being cleaned.
reads.pe.dt {
--cleaning
--clean-by-self:off
--clean-other:off
--stat-base
src.file = "reads1.fa"
src.format = "ifasta-pair" {
def_fasta_qua = 25
phread=64
}
aver_pe=200
stdev_pe=75
}
reads.pe.cl {
--cleaning
--clean-by-self:off
--clean-by-other:off
--clean-other:on
--stat-base
src.file = "reads2.fa"
src.format = "ifasta-pair" {
def_fasta_qua = 25
phread=64
}
aver_pe=200
stdev_pe=75
}
hardware {
memory.opt = 4
memory.max = 8
}
License And Citation
readsClean is a free for academic usage. For commercial licensing, please contact Softberry softberry@softberry.com.