Transomics - pipeline to map RNAseq data, assemble them into transcripts and quantify the abundance of these transcripts in particular datasets.
The pipeline will include the following key components: ReadsMap tools for alignment of RNA-Seq reads to genome, Fgenesh++R identification of alternative transcripts gene prediction tolls, and ExpLevel tool for measuring expression levels of predicted transcript isoforms.
When use please reference: Igor Seledtsov, Vladimir Molodtsov, Peter Kosarev, Victor Solovyev: Transomics pipeline - tools for analysis of RNASeq data. http://linux5.softberry.com/cgi-bin/berry/programs/Transomics
Analysis of transcriptome data proceeds through the following steps:
- makes FASTA files from four-line-format sequence files
- concatenates all *.fa files (from the same set) into one file
- removes head/tail NNNs and skip short reads
- makes text -> binary reads
- maps reads to chromosomes
- sorts reads by chromosomes
- makes a profile - coverage of chromosome by reads
- makes alignments of reads that have no exact mapping (for splice sites discovery)
- makes potential splice sites files
- runs FGENESH gene identification, while accounting for mapped reads in exons and splice sites
- calculates expression values for identified genes using number of mapped reads to a particular gene sequence and the whole genome
Fgenesh++R - gene prediction using RNASeq data
We build the gene identification with RNA-Seq data based on components of FGENESH++ gene finding pipeline (Salamov, Solovyev, 2000; Solovyev et al., 2006; Solovyev, 2007), which is known as one of the most a ccurate eukaryotic gene identification tool and has been used in annotation of a dozen new genomes.
|
|
ReadsMap provide information on splice sites and intron positions that forces modified version of Fgenesh++ to produce gene models that are supported by RNASeq data.
Fgenesh++R provides possibility to predict tissue specific gene variant or even produce alternatively spliced gene models. These models can be visualized in the ReadsMap Viewer.
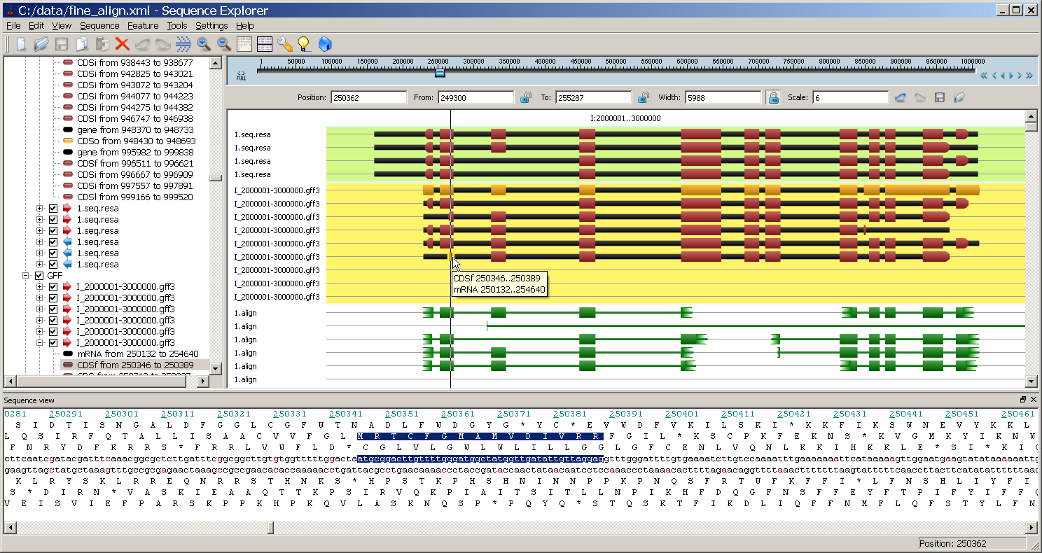
|
Finally, Transomics pipeline includes a module to compute a relative abundance (expression level) of alternative transcripts generated from the same gene locus using a solution of a system of linear equations.

|
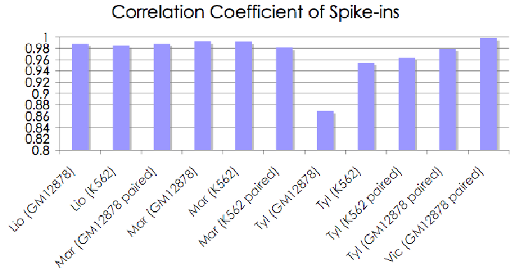
|