TransSeq - Program for assembling transcripts from short reads (RNASeq data)
Our algorithm for contigs assembling comes to clustering of reads.
The algorithm begins with an empty cluster. At the initial step, the first cluster takes the first read from the pool of free reads (that initially contains all the reads). The consensus of the single-read cluster is identical to read sequence. Further, the next read of the pool is to be added to the cluster assuming the alignment of this read to consensus meets certain criteria. Once the second read (as well as all the following ones) is added to the cluster, the consensus is recalculated. All reads included into cluster are excluded from the free reads pool.
To speed up the search for similar sequences in the file with reads, the algorithm utilizes the L-plet (word of length L) method. The algorithm attempts to take only those reads that contain a certain (preliminary defined) number of L-plets identical to terminal fragment of the consensus (of a certain length).
In the case, when there is an information on reads pairness, the cluster is being assembled in two stages. At the first, the cluster is assembled without considering the information on pairs. As soon as contig reaches the length that guarantees the aligning of both reads of a pair, the algorithm starts to check whether reads are mapped to contig in allowed distance. That being the case, both reads of a pair are to be accepted to the cluster and excluded from the pool, and contig is lengthening. If the contig assembling is stopped prior to step that uses the information on pairs, the contig is to be disassembled and reads are to be returned to the pool.
The contig lengthening process continues till the free pool contains reads, aligning of which to consensus allows to include them into current cluster.
The consensus of cluster is output as contig.
The assembling of following clusters (as well as contigs) occurs in the same manner. Clusters are being assembled of the reads that were not used during the assemblage of previous clusters.
The assemblage process is stopped when the pool of free reads is exhausted.
In the case of RNA splicing variants assemblage, the clustering algorithm is featured by a minor difference. Clusters, as for regular algorithm variant, are being initialized with reads of the free pool (that were not used previously). Meanwhile, the reads used for lengthening of a certain contig, in contrast to regular algorithm, can be used further for lengthening of any other contig. Thus, the algorithm mostly guarantees the recovery of any exon, that is covered by a number of reads, as well as adjacent exons.
RAW DATA
Program operates with the array of paired sequences that are organized into file in multifasta format.
Meanwhile, the paired sequences are written to file sequentially (even/odd).
In configuration file for raw sequences the following data are to be specified:
ŕ. Number of sequences in the file (field READS:)
b. Length of sequences (READ_LEN:)
c. Type of pairness (in current version the READ_TYPE:PAIR_ENDS only is supported)
d. Average distance between far-out ends of pairs (LEN_AVER1:)
e. Standard deviation for distance between far-out ends of pairs (LEN_STDEV1:)
The example of configuration file is in the sample.di file
Accuracy evaluation
To evaluate the accuracy we used the artificially created set of sequences.
For these purposes the layout of human chromosome 22 was taken (http://genome.ucsc.edu/, h19 set), and on this basis the file with 1334 mRNA sequences was created.
On the basis of these sequences, pairs of reads with random number of mutations were generated. Thus 1960300 short reads were obtained.
Contigs obtained with use of TransSeq were aligned to original set of mRNAs. Alignment with homology not less than 0.98 and coverage not less than 90% of contig length was considered to be valid. If multiple alignments were obtained, the one with the best score was chosen.
The following attributes were evaluated:
1. Number of found mRNAs - mRNAs for which the corresponding contigs were found
2. Number of found contigs
3. Number of valid contigs - contigs that correspond to original mRNAs
4. Sensitivity (Sn) - ratio of mRNAs number, for which the corresponding contigs were found, to that of original mRNAs
5. Specificity (Sp) - ratio of valid contigs number to that of all found contigs
6. Excessiveness - ratio of found mRNAs number to that of valid contigs
Results
Original number of mRNAs 1334
Number of found mRNAs 1299
Number of found contigs 8511
Number of valid contigs 7789
Number of invalid contigs 722
Sn 0.973
Sp 0.915
Excessiveness = 0,166
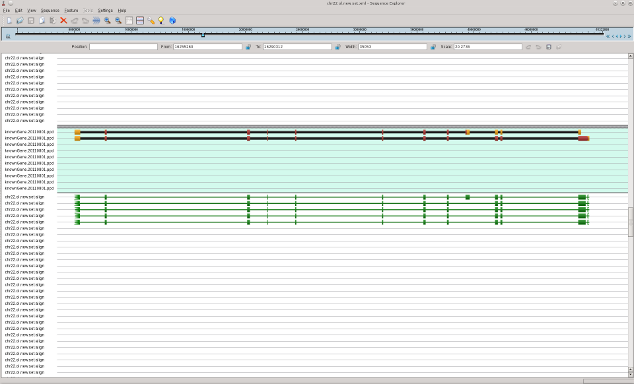
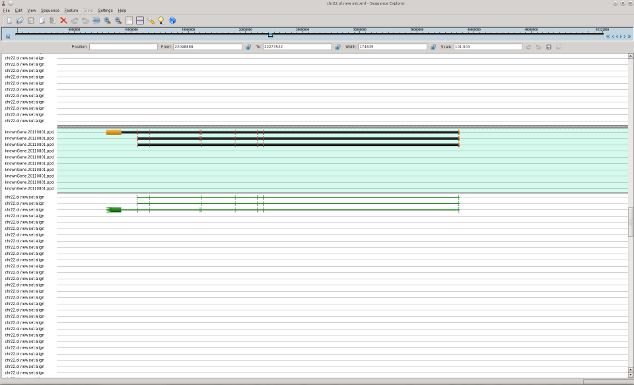
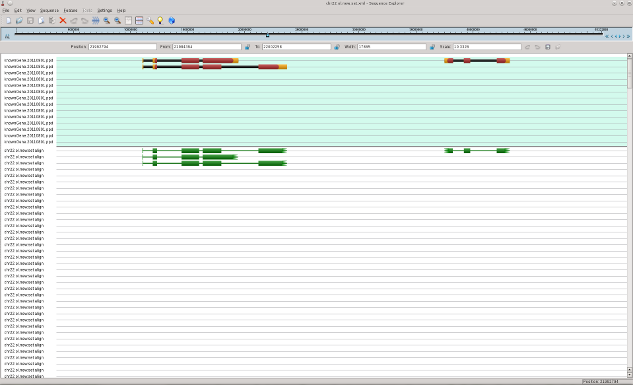
In following pictures the examples of original mRNAs layout (brown color) and results of mapping of obtained contigs to chromosome 22 with use of est_map program are shown.