VirusPoinst
VirusPoints is a software package for detecting viruses integrated in the genome and predicting potential sites integration in a host genome based on NGS data.
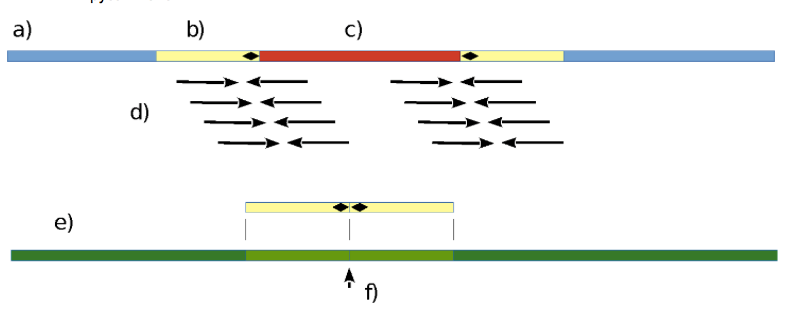
Fig. 1. Schematic comparison of infected and intact genomes.
Assume a genome (a) carrying an integrated virus (c). Using short sequencing reads (d) that cover an integration point (b), it should be possible to deduce a potential integration site (f) in an intact genome (e). Black rhombuses denote virus integration site.
Implementation
In VirusPoints package, we implemented two alternative approaches to finding integration points: Analysis of reads alignment profile (specified by key --regime:profile) and mapping contigs assembled from such reads (specified by key --regime:contigs) to a genome. Both approaches start with sorting reads into the following subsets (Fig. 2):
a) Read pairs completely mapping to a host genome (both reads).
b) Read pairs likewise completely mapping to a viral sequence.
And “border” reads, that extend outside a genome or a viral sequence, which can be divide into two subsets:
c) Pairs where at least one read crosses a border between viral and host sequences and, therefore, contains an integration site of a virus.
d) Pairs where one read completely maps to a host genome and its pair completely maps to a viral genome.
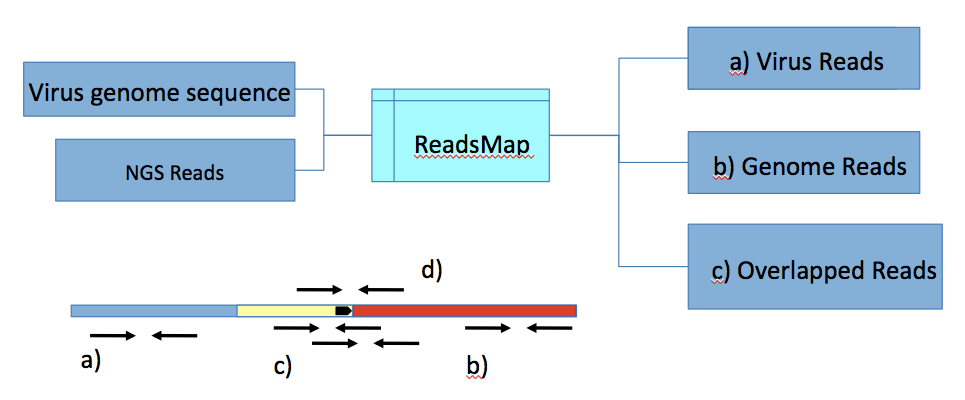
Fig. 2. Sorting reads into subsets.
Viral contig; Initial reads set; (b) Viral reads; (a) (Host) genomic reads; (c), (d) border reads.
We select reads overlapping with a virus genome creating subsets (c) and (d) and discard the reads that map to a genome completely or nearly so, retaining only those which contain sizable stretches of viral sequences. The positions of genome/viral boundaries are saved along with such reads.
Analysis by reads alignment profile
Let’s map a subset of border reads (c) to a host genome without accounting for pairwise reads, and allowing for long unaligned tails. Then a viral integration point would satisfy the following criterion:
There is a defined alignment boundary, while unaligned tails, which are homologous to a viral genome, extend beyond that boundary (Fig. 3). In order to minimize false positives, maximum length of protruding unaligned tails must be set to a value comparable to length of reads (parameter –tailLen:XX), and width of coverage in the integration site shall be comparable with sequencing coverage (parameter —prfTrash:XX).
Such approach has demonstrated to be stable and highly sensitive in case of substantial (>30x) coverage by long (>100 bp) reads.
In case of less substantial coverage, or reads shorter than 100 bp, an approach involving mapping assembled contigs yields superior results.
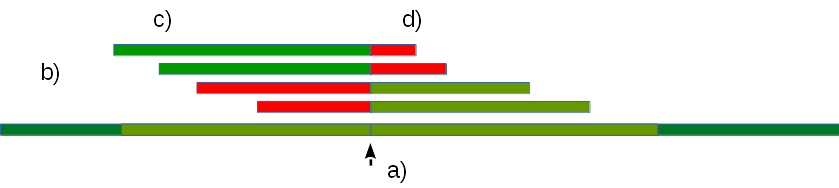
Fig. 3. Mapping reads to a genome. a) — viral integration point; b) reads; с) — portions of reads mapping to host genome (shown in green); d) — portions mapping to viral genome (shown in red).
Analysis using assembled contigs
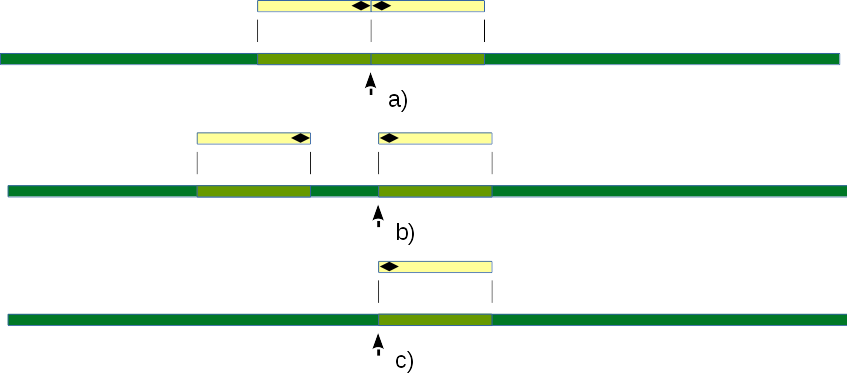
Fig 4. Mapping contigs to a host genome to find viral integration sites.
Testing Performance of VirusPoints –Test on Simulated Viral Insertion Sites
To test performance of the package, we tested it using simulated sequencing reads of human chromosome 20 (hg38) with sequences of a virus totally uncharacteristic to human genome (Stx2 converting phage vB_EcoP_24B) inserted in in eight copies into both strands.
The test reads set consisted of 6,351,716 reads covering both chromosomal and viral sequences. Length of reads was 100 bp, distance between pairwise reads 300, standard deviation 30.
The software has found exact locations of all eight integration sites, confirming their boundaries by mapping reads or contigs from both sides (Fig 5).
VirusPoint 2.01. Search for virus integration position in genome.
Virus file name: Stx2.fa. Sequence 1. >Stx2 converting phage vB_EcoP_24B, complete genome.
Genome file name: chr20_cut.fa.
Reads file name: contigNP.pe.100.300.30.80x.fa.
Pair average distance: 300. Standart deviation 30.
Both sides supported positions:
1 Pos: 69251 | From left L: 75 | From right L: 50 | Support: 43 | Sequence: 1 Name:chr20
2 Pos: 118601 | From left L: 74 | From right L: 51 | Support: 46 | Sequence: 1 Name:chr20
3 Pos: 155101 | From left L: 74 | From right L: 49 | Support: 37 | Sequence: 1 Name:chr20
4 Pos: 514601 | From left L: 71 | From right L: 49 | Support: 53 | Sequence: 1 Name:chr20
5 Pos: 6810801 | From left L: 74 | From right L: 51 | Support: 38 | Sequence: 1 Name:chr20
6 Pos: 7137501 | From left L: 75 | From right L: 50 | Support: 37 | Sequence: 1 Name:chr20
7 Pos: 7455951 | From left L: 71 | From right L: 50 | Support: 40 | Sequence: 1 Name:chr20
8 Pos: 7485651 | From left L: 74 | From right L: 50 | Support: 30 | Sequence: 1 Name:chr20
-----------------------------------------
Fig 5. Example of output on a simulated example.
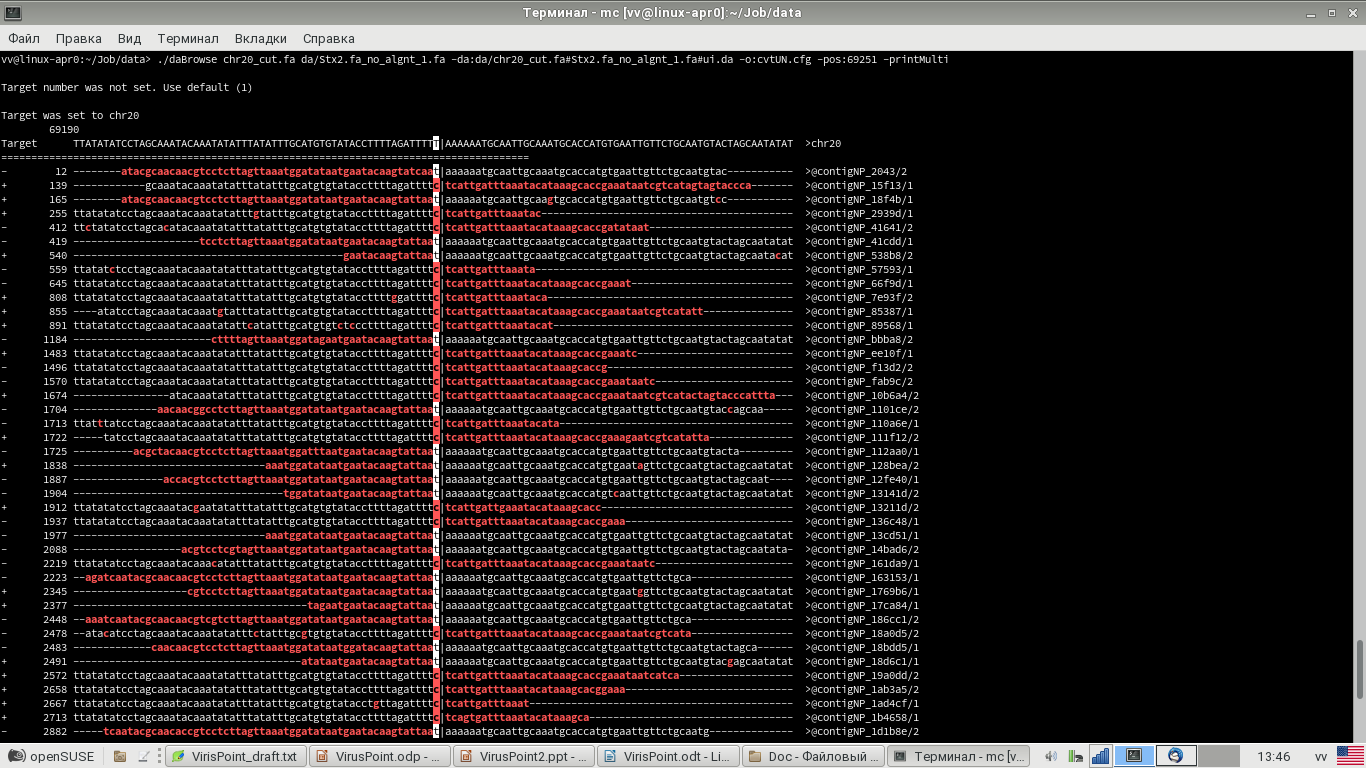
Fig 6. Example of output of alignments of reads to a genome around viral integration site (unaligned viral tails are shown in red) – compare with Fig. 3.
Example of Use — Real World Data
The software was also tested on real world reads (reads set CaSki - Viral, clinical or host-associated sample from human papillomavirus).
The block for identifying viral sequences, built into Virus Point software, has determined that the sequence of papillomavirus present in sequencing reads most likely belongs to gi_3377787_gb_U89348.1_HPU89348 Human papillomavirus type 16 variant.
The software has found one exact site and three regions of viral integration (Fig 7).
VirusPoint 2.01. Search for virus integration position in genome.
Virus file name: 37.fa. Sequence 1. >gi_3377787_gb_U89348.1_HPU89348_Human_papillomavirus_type_16_variant,_complete_s.
Genome file name: chr_hg38.fa.
Reads file name: Caski.fa.
Pair average distance: 200. Standart deviation 65.
Both sides supported positions:
1 Pos: 11700450 | From left L: 122 | From right L: 103 | Support: 322 | Sequence: 1 Name:chr10
-----------------------------------------
Regions with unknown exact positions:
1 Reg: 80864908 - 80865993 | From left L: 121 | From right L: 103 | Support: 116 | Sequence: 2 Name:chr11
2 Reg: 100771862 - 100766769 | From left L: 116 | From right L: 101 | Support: 227 | Sequence: 2 Name:chr11
3 Reg: 145716515 - 145708232 | From left L: 122 | From right L: 102 | Support: 294 | Sequence: 23 Name:chrX
-----------------------------------------
Positions with partial support:
Head
1 Pos: 100807257 | L: 120 | Support: 67 | Sequence: 2 Name: chr11
2 Pos: 100819015 | L: 107 | Support: 49 | Sequence: 2 Name: chr11
3 Pos: 55310221 | L: 126 | Support: 91 | Sequence: 10 Name: chr19
4 Pos: 56203737 | L: 113 | Support: 42 | Sequence: 10 Name: chr19
5 Pos: 7565064 | L: 119 | Support: 33 | Sequence: 13 Name: chr21
6 Pos: 20582476 | L: 122 | Support: 33 | Sequence: 13 Name: chr21
7 Pos: 22416219 | L: 109 | Support: 101 | Sequence: 15 Name: chr2
8 Pos: 22422772 | L: 122 | Support: 227 | Sequence: 15 Name: chr2
9 Pos: 22479211 | L: 124 | Support: 31 | Sequence: 15 Name: chr2
10 Pos: 27136176 | L: 123 | Support: 180 | Sequence: 15 Name: chr2
11 Pos: 46292456 | L: 115 | Support: 44 | Sequence: 18 Name: chr5
12 Pos: 45691386 | L: 122 | Support: 250 | Sequence: 19 Name: chr6
Tail
Tail
1 Pos: 6741036 | L: 106 | Support: 217 | Sequence: 2 Name: chr11
2 Pos: 67786650 | L: 104 | Support: 34 | Sequence: 2 Name: chr11
3 Pos: 42967878 | L: 108 | Support: 34 | Sequence: 5 Name: chr14
4 Pos: 54971194 | L: 108 | Support: 34 | Sequence: 10 Name: chr19
5 Pos: 55307400 | L: 100 | Support: 333 | Sequence: 10 Name: chr19
6 Pos: 26276704 | L: 101 | Support: 301 | Sequence: 12 Name: chr20
7 Pos: 26358220 | L: 101 | Support: 64 | Sequence: 12 Name: chr20
8 Pos: 5759414 | L: 102 | Support: 33 | Sequence: 13 Name: chr21
9 Pos: 5879998 | L: 102 | Support: 33 | Sequence: 13 Name: chr21
10 Pos: 8157484 | L: 102 | Support: 33 | Sequence: 13 Name: chr21
11 Pos: 8340529 | L: 102 | Support: 33 | Sequence: 13 Name: chr21
12 Pos: 22416232 | L: 103 | Support: 408 | Sequence: 15 Name: chr2
13 Pos: 44893594 | L: 108 | Support: 34 | Sequence: 19 Name: chr6
14 Pos: 45652126 | L: 105 | Support: 60 | Sequence: 19 Name: chr6
15 Pos: 45691418 | L: 101 | Support: 100 | Sequence: 19 Name: chr6
16 Pos: 6925193 | L: 104 | Support: 117 | Sequence: 20 Name: chr7
17 Pos: 54076318 | L: 101 | Support: 64 | Sequence: 20 Name: chr7
18 Pos: 63523952 | L: 100 | Support: 39 | Sequence: 20 Name: chr7
-----------------------------------------
Pos: - position on chromosome.
Reg: - region on chromosome.
L: - chromosome alignment length.
Fig 7 — The results of search by Viral Point for integration points of human papillomavirus type 16 variant (CaSki reads) into human genome (hg38).