Advanced suit of robust bioinformatics tools for efficient analysis of large-scale NGS data
Dozens new algorithms have been developed for next-generation sequencing (NGS) data processing. The collaborative competitions
(such as Assemblathon, Alignathon and RGASP) assessed the state of the art in genome assembling, read mapping,
discovery alternative transcripts. They demonstrated the lack of consistency between software tools in terms of comparisons
obtained on different data sets as well as relative to various metrics evaluating the quality of results suggesting
that there is still much room for improvement. We developed an advanced suit of robust bioinformatics tools for efficient
analysis of large-scale NGS data. The OligoZip algorithm is enhanced by iterative module of accounting paired reads and adding a new procedure
of restoration of repeated sequences in assembled scaffolds. It includes
- OligoZip -
de novo NGS reads assembler. Softberry OligoZip tool for processing short reads from next-generation sequencing machines, such as Solexa/Illumina and similar, provides effective solutions to the following tasks:
-
De novo reconstruction of genomic sequence;
-
Reconstruction of sequences based on reference genome from same or close species;
-
Mutation profiling and SNP discovery in a given set of genes;
-
Analysis of transcriptome sequence data with estimates of gene expression level and identification of gene structure of expressed splice variant.
Test online OligoZip.
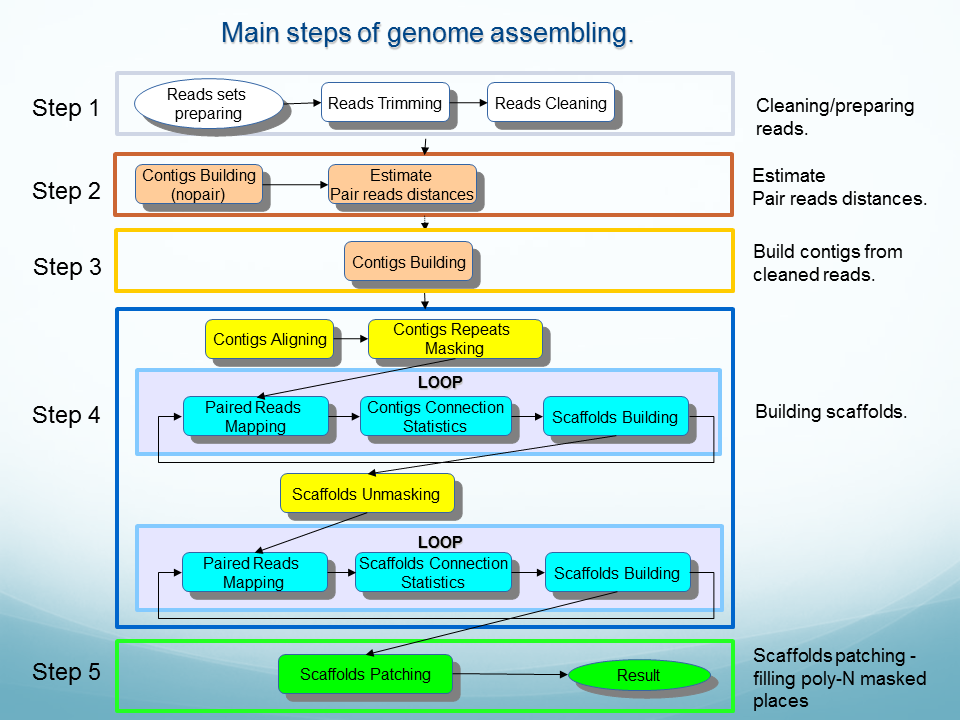
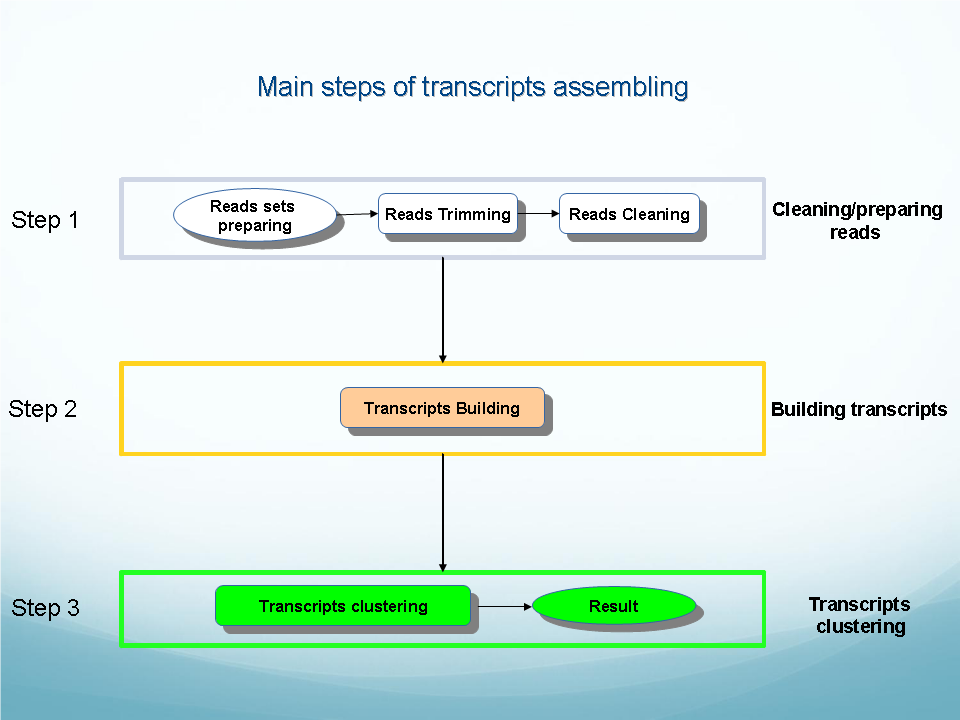
See descriptions of OligoZip variants: OligozipDNA ( see flowchart) and OligoZipRNA ( see flowchart).
- ReadsMap -
ReadsMap is a fast short read aligner that quickly maps/aligns large sets of short DNA sequences. Multiple processors can be used optionally to achieve greater alignment speed. On initial stage we map “exonic” reads that demonstrate high-quality, non-interrupted alignment to a genomic sequence. Potentially, this step would map most of the reads to a genome, and the remaining “non-mapped” group would be small enough to be subjected to more thorough analysis. At the second step, we use a modified variant of our EST_MAP program to align these “non-mapped” reads using splice site matrices and producing very accurate alignment with gaps. This reads will indentify potential exon-intron boundaries.
ReadsMap has been significantly improved in the speed of data processing and its accuracy to map RNASeq reads
reached Sensitivity 0.99 and Specificity 0.96. RNASeq reads clustering program that assembles the RNA-Seq data
into unique sequences of transcripts generates full-length transcripts for a set of alternatively spliced isoforms.
The program demonstrates Sensitivity 0.97 and Specificity 0.96 in identifying known RNA transcripts on test data.
Test online ReadsMap
See descriptions of ReadsMap (both RNASeq and Genomic (no introns) variants).
- TransSeq -
program for de novo assembling alternative transcripts from short reads and gene expression quantification.
Test online TransSeq
-
GenomeMatch - a tool to compare genomic sequences. The pipeline job result is an optimal (most full with maximal homology)
coverage of every "target" sequences by fragments of the "query" set ones.
Test online GenomeMatch
- Transomics pipeline -
The pipeline will include the following key components: ReadsMap tools for alignment of RNA-Seq reads to genome, Fgenesh++R identification of alternative transcripts gene prediction tolls, and ExpLevel tool for measuring expression levels of predicted transcript isoforms.
Test online Transomics
-
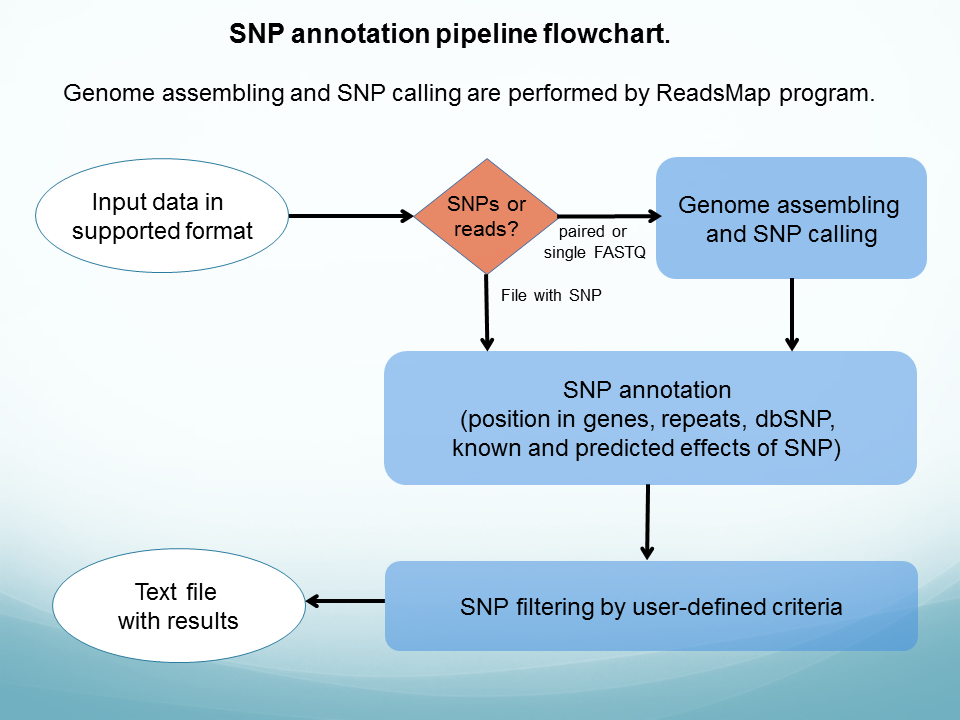
SNP discovery and annotation pipeline ( see flowchart) - annotates a set of human SNPs.
-
Constructing and Visualizing synteny for assembled genomes.
{kind=link}
{kind=link}
{kind=link}