Main Products
Below is a partial list of Softberry products. Most programs that are available for online testing can also be licensed, even if they are not in this list. Complete list of products is available upon request. We license our products on subscription basis, with licensing fees payable annually. Typically, we offer only two types of licenses: personal single-user and site license. Fees for academic single-user licenses vary from several hundred to a few thousand dollars per year. For instance, such license to EST_Map or Prot_Map is $1,000 per year, FGENESH - $1,640 for the first year and $1,120 annual renewals etc. Site licenses usually cost 2.5x as much. Commercial licenses are obviously more expensive: please inquire for more details. Most of our programs are available for major Unix-derived platforms, such as Linux, Mac OSX and Cygwin environment under Windows.
-
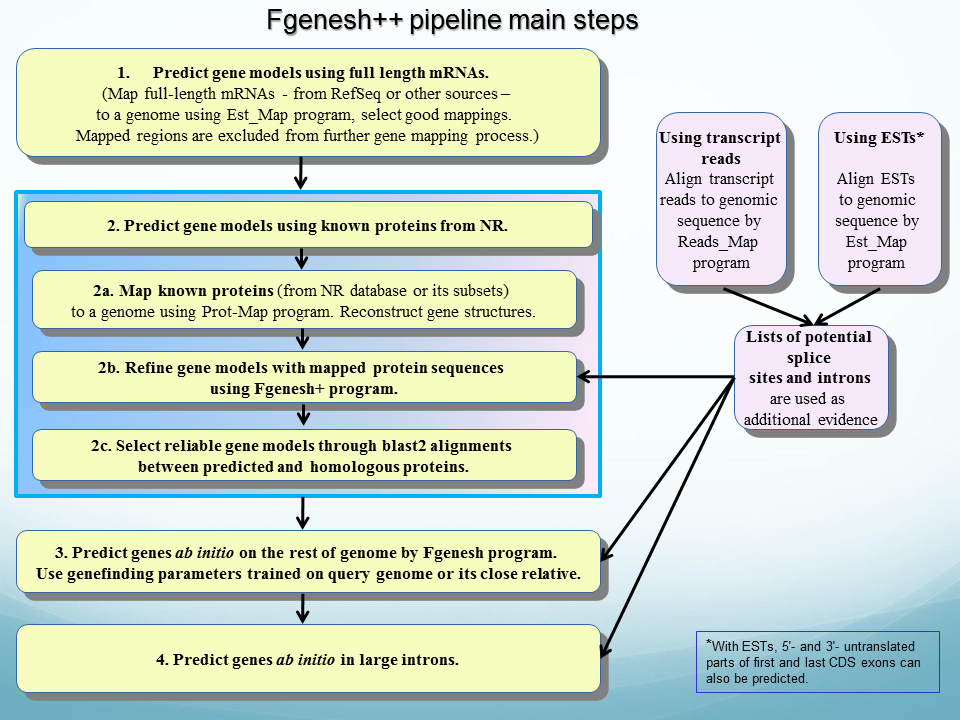
Eukaryotic genome annotation pipeline Fgenesh++ (see flowchart) that includes the following steps:
-
a) Mapping known genes/mRNAs from RefSeq or other sources (EST_Map program);
-
b) Mapping known proteins (typically from NR database) to a genome;
-
c) Identification of splice sites by mapping a set of known ESTs or NGS RNASeq reads;
-
d) Identification of gene models that involve alternative splicing.
See examples in Sequence annotation explorer.
-
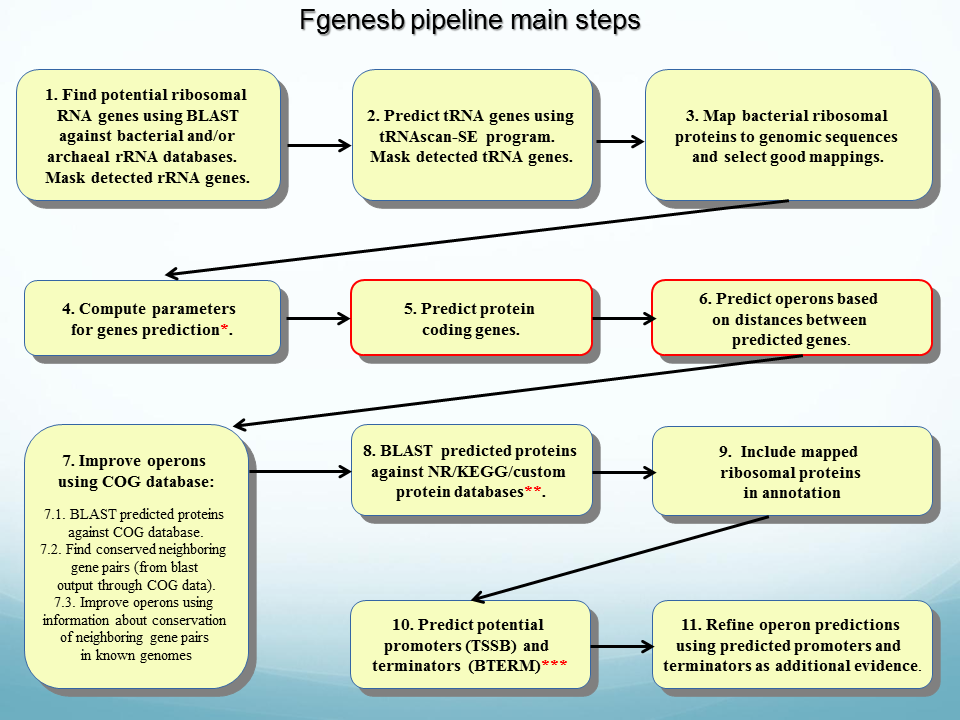
Bacterial genome annotation pipeline Fgenesb ( see flowchart) predicts genes (including RNA genes), operons, promoters and terminators. It provides functional and pathway annotation of predicted proteins with the user selected databases such as NR, COGS, EggNOG, KEGG and etc.
-
Eukaryotic and Bacterial genes and functional sites identification and genome annotation:
-
Fgenesh: the fastest ab initio HMM-based eukaryotic
gene prediction program with parameters for
506 genomes.
-
Other programs of FGENESH suite include
FGENESH-2,
FGENESH+ and
FGENESH_C,
which perform genefinding with support of related genomic, or protein or cDNA sequence, respectively, programs for Best ORF and splice sites, non-standard splice sites identification.
-
BESTORF - finding potential coding fragment EST/mRNA
-
FSPLICE - finding splice sites in genomic DNA
-
FEX - finding potential 5'-, internal and 3'-coding exons
-
BPROM - prediction of bacterial promoters
-
FindTerm - Finding Terminators in bacterial genomes
-
Analysis of NGS data: genome and transcriptome assembling, reads mapping, SNP discovery and evaluation:
-
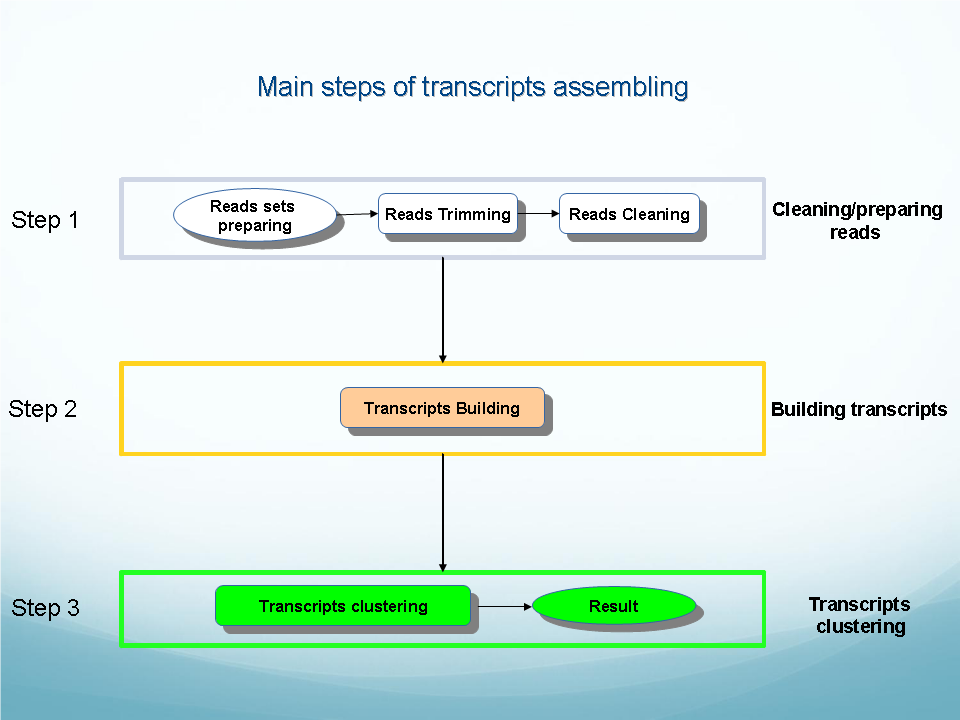
OligozipDNA ( see flowchart): Program for sequence assembly that includes error correction, contig and scaffold building.
-
ReadsMap: Program for mapping genomic and RNASeq reads to a genome, with identification of splice sites and SNPs.
-
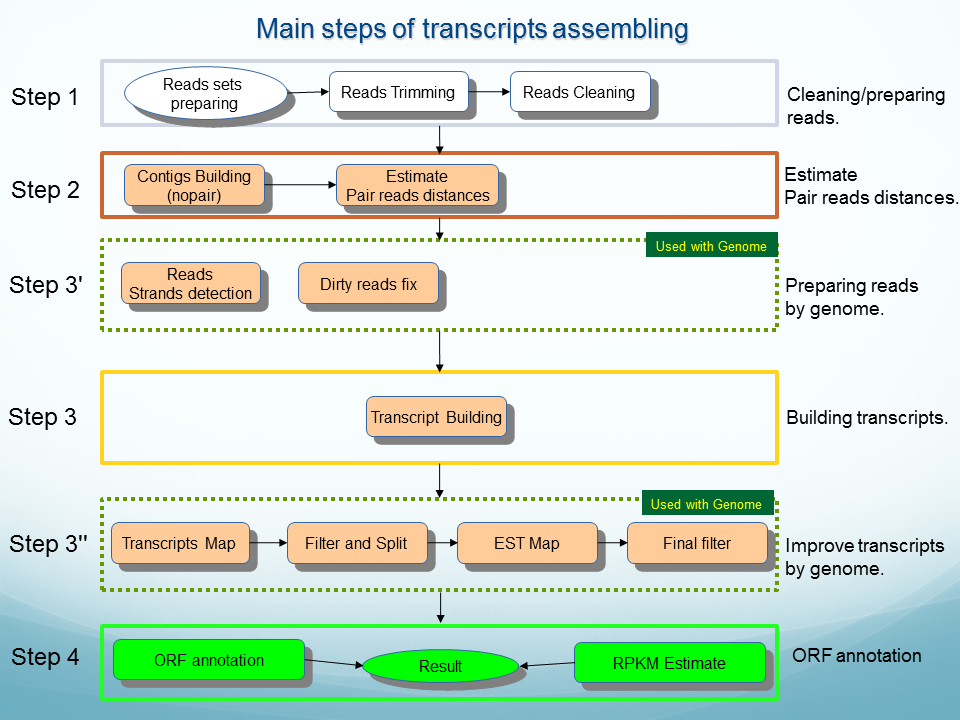
OligoZipRNA ( see flowchart): Program for assembling RNASeq reads to RNA transcripts, selection of alternative transcript groups and identification of RNA and protein coding subsets.
See examples in Sequence annotation explorer.
-
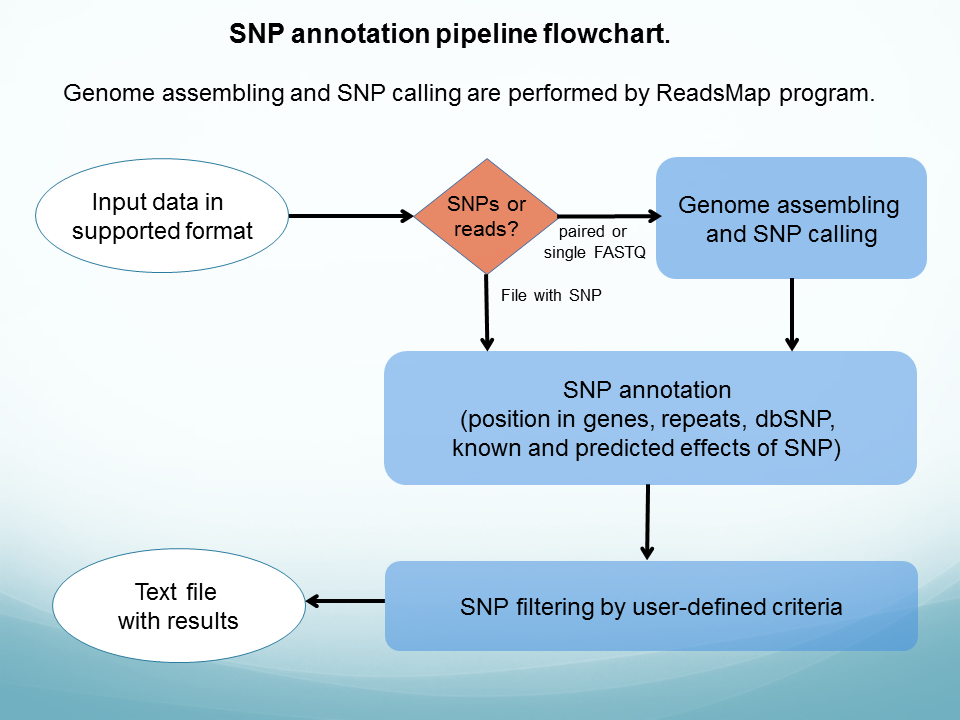
SNP discovery and annotation pipeline ( see flowchart).
- TransSeq -
program for de novo assembling alternative transcripts from short reads and gene expression quantification.
The algorithm begins with an empty cluster. At the initial step, the first cluster takes the first read from the pool of free reads (that initially contains all the reads). The consensus of the single-read cluster is identical to read sequence. Further, the next read of the pool is to be added to the cluster assuming the alignment of this read to consensus meets certain criteria. Once the second read (as well as all the following ones) is added to the cluster, the consensus is recalculated. All reads included into cluster are excluded from the free reads pool.
Test online TransSeq
- Transomics pipeline -
The pipeline will include the following key components: ReadsMap tools for alignment of RNA-Seq reads to genome, Fgenesh++R identification of alternative transcripts gene prediction tolls, and ExpLevel tool for measuring expression levels of predicted transcript isoforms.
Test online Transomics
-
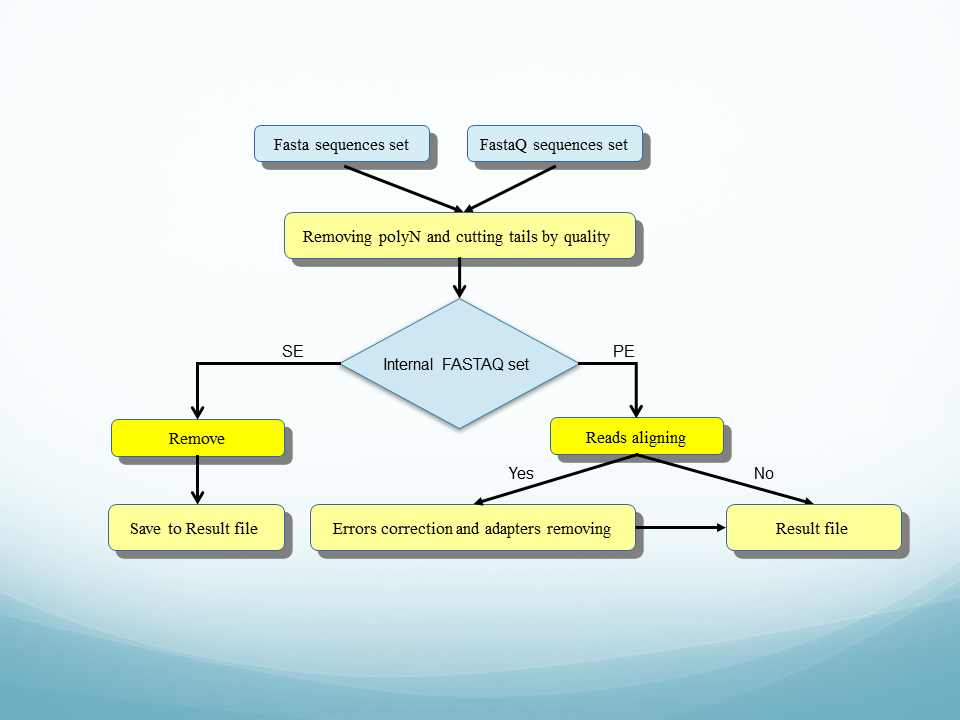
Adapter_trim ( see flowchart) is a newest program for the preparation of short sequences (reads) sets for further analysis. The adapter_trim main goal is removing adapters from reads, but you can use it for the task of searching and removing polyN tails and cutting sequence by quality.
-
Constructing and Visualizing synteny for assembled genomes.
-
Promoter and functional sites prediction:
-
TSSW/
TSSP/
TSSB: Programs for predicting animal, plant and bacterial promoters and functional sites.
-
RegSite: Database of gene regulatory elements.
-
Nsite/
NsiteM/
NsiteH: programs for analysis of regulatory regions and composition of their functional motifs. .
-
POLYAH: - Recognition of 3'-end cleavage and polyadenilation region
-
PATTERN,
ScanWM-PL : search for patterns.
-
CpGFinder: GC-islands finding .
-
Database search, genome comparison and Protein/RNA/EST/ mapping to genome programs
-
PROT_MAP: mapping of a set of proteins on genome.
-
EST_map: Mapping your mRNA/EST to Chromosome sequence of Human genome
-
SMAP: mapping oligonucleotides to genome.
-
DBSCAN :database homology search program similar to BLAST, but handling multimegabyte-size sequences.
-
Protein function and structure prediction and analysis
-
ProtComp: Program for predicting protein sub-cellular location, works on all taxons, including animals, plants, fungi and bacteria.
-
PDISORDER: Protein Disorder Prediction.
-
CYS_REC,
PSSFinder,
SSP
NNSSP
SSPRED
SSENVID
SSPAL:
programs of secondary structure prediction.
-
HMod3DMM: energy minimization program by molecular mechanic.
-
3DmodelFit: program for comparison protein 3D model and its original structure.
-
MDynSB: Optimization of a protein structure via MD simulation in an implicit water solvent, optimization and folding of a protein via (the user defined) simulated annealing protocol in an implicit water solvent, optimization of a predefined protein loops while non-loop parts of the protein molecule is kept fixed in the course of the loop optimization.
-
3D-Match, 3D-MatchDB and etc: Programs for protein structure comparison, prediction and analysis.
See examples in 3D-explorer.
-
Alignment genome sequences, synteny reconstruction
-
RNA structure computing
-
Seqman - manipulation with sequences
-
SelTag - a tool for analysis of expression data
-
MolQuest: Comprehensive, easy to use desktop application for sequence analysis. Includes ~ 100 program modules, dozen viewers and GUI running on Window, Linux and Mac OS platforms.
See
www.molquest.com for details.
-
Proteomics - MS data processing.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}